Two very interesting new articles have been published on the (supposed) action of a drug against Alzheimer's disease.
 These articles provide the reader with several points for further reflection.
- One of them is that the clinical trial cannot be the only method of validating the marketing of a drug. Indeed, for diseases that develop over decades, often asymptomatically, how can the effectiveness of a drug be tested? We can always dream of a pill that will cure in a few months a patient, as this is true for diseases where there is an identified pathogen, but it is very unlikely with chronic diseases. Even for communicable diseases, this type of medication does not always exist; a patient can be considered permanently cured, even though they suffer very serious after-effects.
These articles provide the reader with several points for further reflection.
- One of them is that the clinical trial cannot be the only method of validating the marketing of a drug. Indeed, for diseases that develop over decades, often asymptomatically, how can the effectiveness of a drug be tested? We can always dream of a pill that will cure in a few months a patient, as this is true for diseases where there is an identified pathogen, but it is very unlikely with chronic diseases. Even for communicable diseases, this type of medication does not always exist; a patient can be considered permanently cured, even though they suffer very serious after-effects.
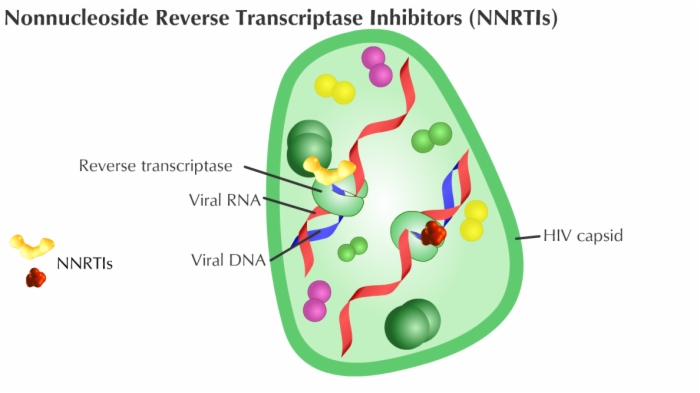
HIV and many other viruses rely on an enzyme, called reverse transcriptase (RT), to copy their RNA molecules and change them into complementary DNA duplicates that can then be inserted back into the host cell's DNA, producing permanent sequence changes.
As RT hijacks a host's cells to establish a chronic infection, so drugs that block the RT enzyme's activity have become a common part of treatment cocktails for keeping HIV at bay.
The team of the first article analyzed anonymized medical records with prescription claims from more than 225,000 controls and patients and found that RT inhibitor exposure was associated with a statistically significant reduced incidence and prevalence of Alzheimer's disease. There were 2.46 Alzheimer's disease diagnoses per 1,000 persons taking these inhibitors, versus 6.15 for the general population. And this with a drug that is not optimized for Alzheimer's disease. Indeed the drugs patients took in this retrospective study were designed to counter a specific form of RT used by HIV and it may only have a limited effect on many different possible forms of the enzyme in the brain. So there is significant room for progress to perfect this drug.
- Another point of reflection concerns the etiology of Alzheimer's disease. This is the subject of numerous debates and gigantic financial bets. Clearly, the pharmaceutical industry ecosystem considers that the appearance of intra and extra-cellular beta-amyloid plaques is the cause of this disease. Few scientists question why these plaques appear, or whether competing hypotheses have been properly studied. In this new article, it is shown that an anti-viral drug significantly reduces the risk of developing Alzheimer's disease.
In a second article, an explanation is proposed as to why clinical trials for this disease most often fail. The amyloid hypothesis, or the theory that the accumulation of a protein called beta-amyloid in the brain causes Alzheimer's disease, has driven Alzheimer's research to date. However, treatments that target beta-amyloid have notoriously failed in clinical trials. The authors of the second article found that most Alzheimer's disease brain samples contained an over-abundance of distinct APP gene variants, compared to samples from normal brains. Among these Alzheimer's-enriched variations, the scientists identified 11 single-nucleotide changes identical to known mutations in familial Alzheimer's disease—a very rare inherited form of the disorder. Although found in a mosaic pattern, identical APP variants were observed in the most common form of Alzheimer's disease, further linking gene recombination in neurons to disease.
“The thousands of APP gene variations in Alzheimer's disease provide a possible explanation for the failures of more than 400 clinical trials targeting single forms of beta-amyloid or involved enzymes,” says Chun. "APP gene recombination in Alzheimer's disease may be producing many other genotoxic changes as well as disease-related proteins that were therapeutically missed in prior clinical trials. The functions of APP and beta-amyloid that are central to the amyloid hypothesis can now be re-evaluated in light of our gene recombination discovery."
- We have all learned that all the cells of an organism have the same genetic heritage since they come from the same original cell. This vision, which dates from the middle of the last century, is beginning to be called into question in different cases: cancers, cases of mosaicism, and aging. These two articles, which come from the same laboratory, raise the question of how to detect these cases of mosaicism. Indeed, if a virus modifies the genome of a cell, but only in certain tissues (such as the brain) and only for a fraction of the cells, a genetic analysis of an easily accessible tissue will not necessarily highlight an alteration of the genome. of a significant fraction of the cell population of another tissue.
The researchers note that: "It is important to note that none of this work would have been possible without the altruistic generosity of brain donors and their loving families, to whom we are most grateful. Their generosity is yielding fundamental insights into the brain and is leading us toward developing new and effective ways of treating Alzheimer's disease and possibly other brain disorders—potentially helping millions of people.
As usual, it is not the first time that RT inhibitors have been proposed against Alzheimer's disease animal models, but it is the first time the effect has been shown in humans.
 The exact reasons why atrial fibrillation (a common heart disease) increases dementia risk aren't fully understood, but there are likely several factors involved. Some of these factors directly cause damage, while others increase the chances of getting dementia over time. These factors include tiny blood blockages and bleeding in the brain's small blood vessels. Small strokes you might not even notice, reduce blood flow to the brain, increase long-term inflammation, and lead to shrinkage of brain tissue.
The exact reasons why atrial fibrillation (a common heart disease) increases dementia risk aren't fully understood, but there are likely several factors involved. Some of these factors directly cause damage, while others increase the chances of getting dementia over time. These factors include tiny blood blockages and bleeding in the brain's small blood vessels. Small strokes you might not even notice, reduce blood flow to the brain, increase long-term inflammation, and lead to shrinkage of brain tissue.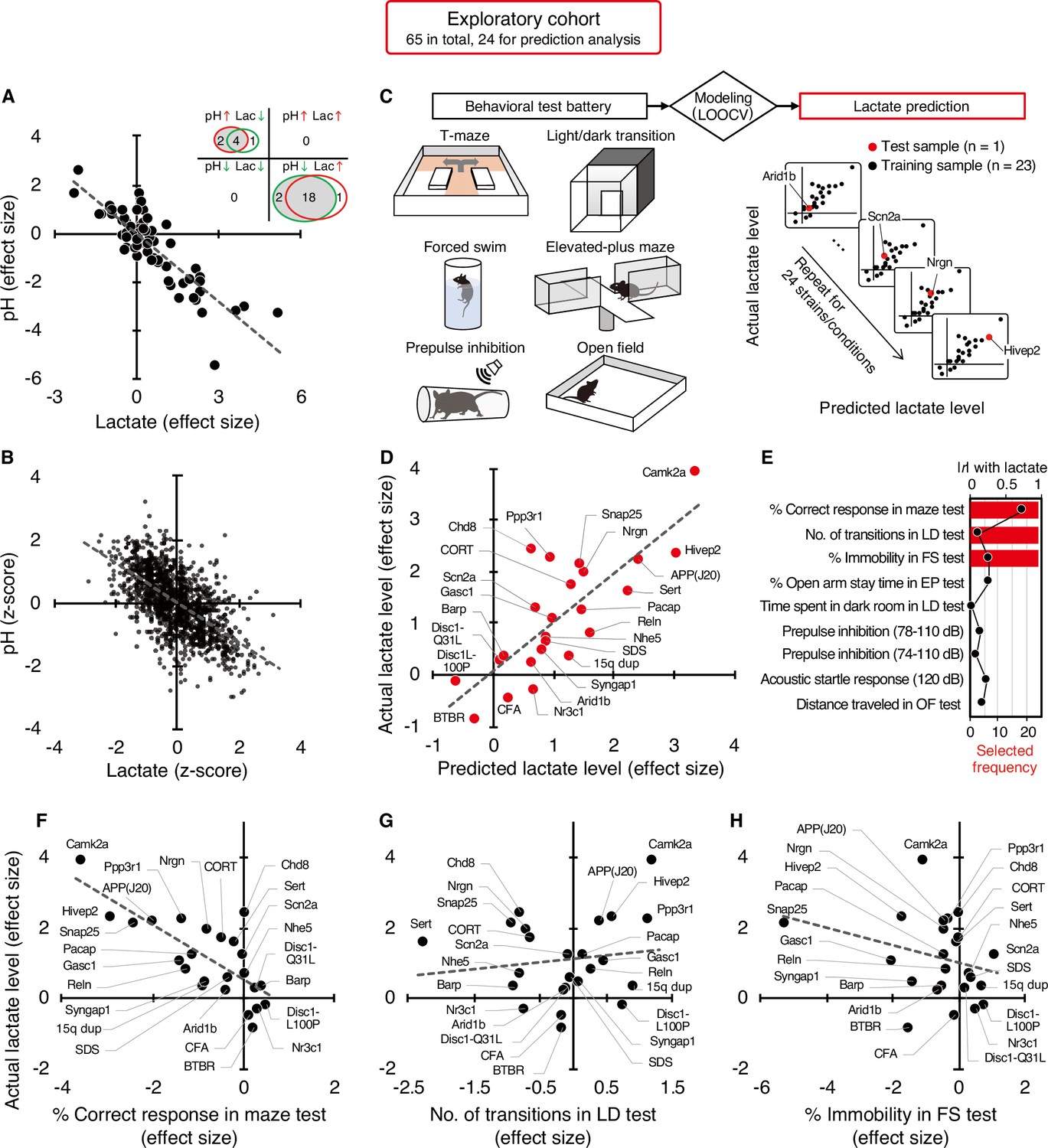 What could be the mechanism between increased lactate and decreased pH?
Lactate is a byproduct of metabolism and it is an acid, hence the acidification of pH. Neuronal activity relies heavily on glucose for energy. Under normal conditions, oxygen is readily available, and glucose is broken down through aerobic respiration, producing ATP (energy) with minimal lactate as a byproduct. However, during periods of high neuronal activity or limited oxygen supply, cells shift to anaerobic metabolism, relying more on glycolysis. This process produces more lactate as a byproduct.
What could be the mechanism between increased lactate and decreased pH?
Lactate is a byproduct of metabolism and it is an acid, hence the acidification of pH. Neuronal activity relies heavily on glucose for energy. Under normal conditions, oxygen is readily available, and glucose is broken down through aerobic respiration, producing ATP (energy) with minimal lactate as a byproduct. However, during periods of high neuronal activity or limited oxygen supply, cells shift to anaerobic metabolism, relying more on glycolysis. This process produces more lactate as a byproduct. Improving cognition using a simple probiotic would be an extraordinary result due to its simplicity of implementation.
Improving cognition using a simple probiotic would be an extraordinary result due to its simplicity of implementation.
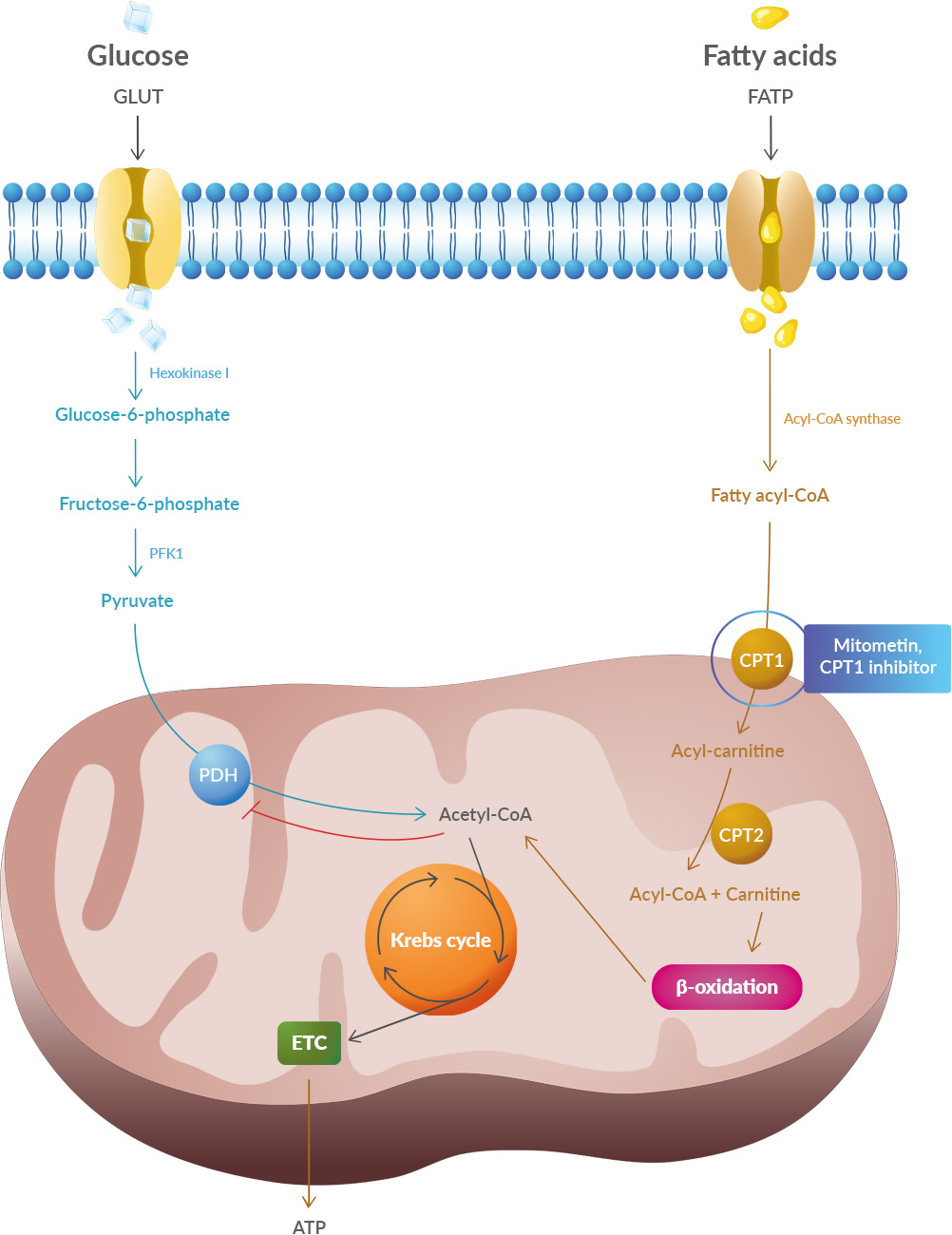 Studies have shown that oxidative stress and endoplasmic reticulum stress are correlated and can lead to protein misfolding (Abramov et al., 2020). Accumulation of misfolded proteins causes cellular damage and mitochondrial dysfunction and is associated with a range of neurodegenerative diseases, including ALS (misfolded SOD1, TDP-43, C9orf72) (McAlary et al., 2020), Parkinson's disease (misfolded α-synuclein) and Alzheimer disease (misfolded Aβ and Tau) (Abramov et al., 2020).
Studies have shown that oxidative stress and endoplasmic reticulum stress are correlated and can lead to protein misfolding (Abramov et al., 2020). Accumulation of misfolded proteins causes cellular damage and mitochondrial dysfunction and is associated with a range of neurodegenerative diseases, including ALS (misfolded SOD1, TDP-43, C9orf72) (McAlary et al., 2020), Parkinson's disease (misfolded α-synuclein) and Alzheimer disease (misfolded Aβ and Tau) (Abramov et al., 2020).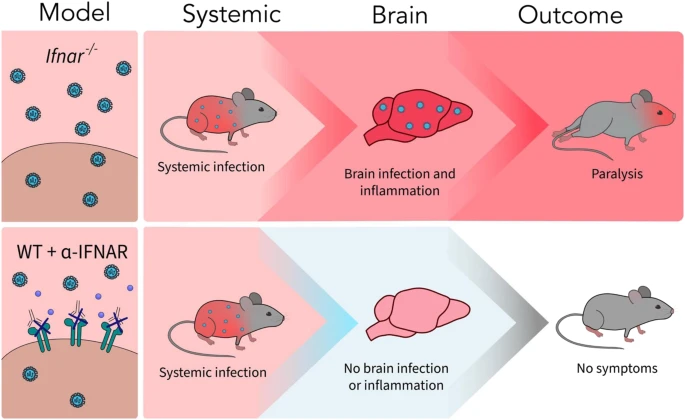 A new article aims to show that in the case of Zika viruses,
A new article aims to show that in the case of Zika viruses, 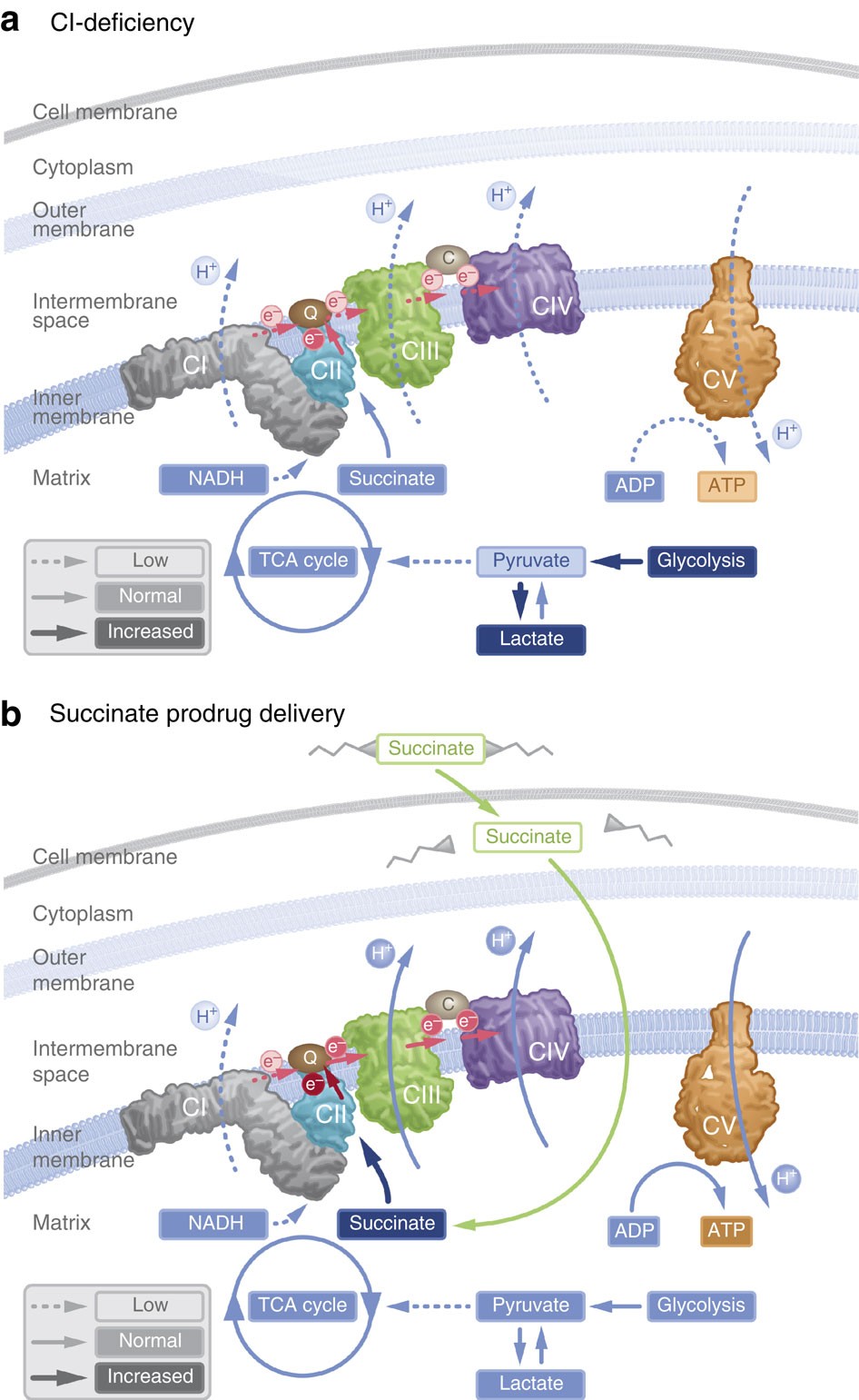 In particular, supplementation with dimethyl succinate, a substrate bypassing the inhibited step in the TCA cycle, suggests a potential therapeutic strategy to mitigate mitochondrial dysfunction in Alzheimer's disease. Dimethyl succinate (DMS), a membrane-permeant form of succinate, could serve as a pro-drug to provide substrate to the next enzymatic step in the TCA cycle, succinate dehydrogenase (SDH).
In particular, supplementation with dimethyl succinate, a substrate bypassing the inhibited step in the TCA cycle, suggests a potential therapeutic strategy to mitigate mitochondrial dysfunction in Alzheimer's disease. Dimethyl succinate (DMS), a membrane-permeant form of succinate, could serve as a pro-drug to provide substrate to the next enzymatic step in the TCA cycle, succinate dehydrogenase (SDH).