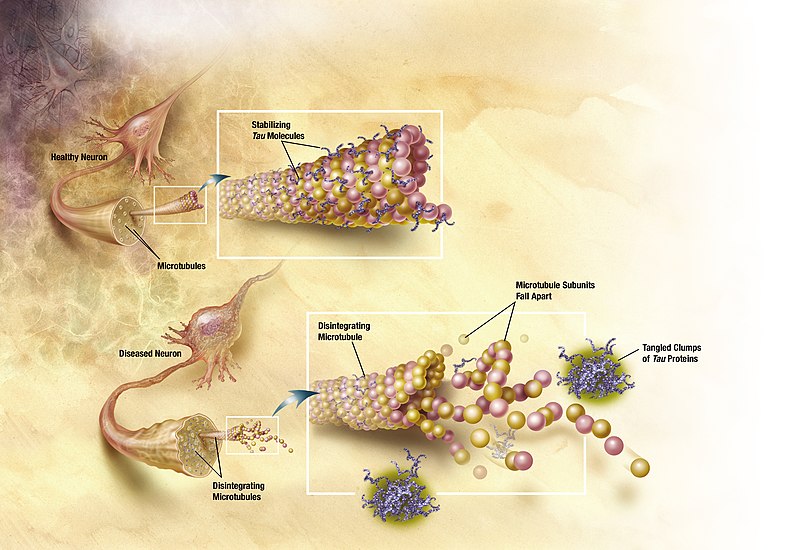
The end of the amyloid plaque hypothesis for Alzheimer's disease
Nearly two decades have passed since an Alzheimer's drug was approved. This, too, is reminiscent of the situation of ALS.
Researchers working on Alzheimer's disease, have broadened their approaches after years devoted to the so-called amyloid hypothesis, which tells that the accumulation of a protein called beta-amyloid in brain plaques is at the origin of the disease. Indeed the tests of drugs that had to eliminate these plaques, have all regularly failed in patients. A similar situation is true also in ALS and also for other neurodegenerative diseases. Maybe our basic concepts about these diseases are wrong.
In October 2019, however, Biogen announced that it would seek approval by the Food and Drug Administration for an anti-amyloid drug called aducanumab, which had failed in two previous trials. The company said a new analysis of some of the trial's data had shown a reduction in cognitive decline in some patients with early Alzheimer's disease.
The intestinal microbiota
The gut microbiota changes before the onset of Alzheimer's disease and these alterations can be detected earlier at the stage of mild cognitive impairment.
There is a certain relationship between amyloid plaques and certain microbes. Several bacterial species, including Bacillus subtilis, E. coli, Mycobacterium tuberculosis, Salmonella enterica, Salmonella typhimurium and Staphylococcus aureus, are capable of producing amyloid fibers. Everything goes as if instead of being a perfect body, in accordance with school books, the aging human body was loaded with years of parasites and various debris in the manner of an old boat hull.
Previous studies have thus shown a correlation between the genera Escherichia, Blautia, Bifidobacterium, Streptococcus, Lactobacillus and Dorea and the appearance of Alzheimer's disease.
On the other hand, the genus Bacteroides could be one of the protective factors of the microbiota. Indeed in the same studies, patients with Alzheimer's disease and mild cognitive impairment show a decrease in the number of Bacteroides, which could be explained by its ability to protect the intestinal barrier.
In September, a group of Chinese scientists, some of whom were employed by the Green Valley Society, published a study in Cell Research that highlighted the link in the mouse between an impaired intestinal microbiome and neuroinflammation.
They reported that oligomannate suppresses bacterial imbalance in the digestive system of mice and reverses cognition disorders. They suggest a new strategy for Alzheimer's treatments by remodeling the intestinal microbiota. However caution was needed because thousands of experimental therapies have cured a considerable number of diseases in mice without ever showing any benefit in humans, but the results of the Phase III clinical trial are changing the game.
Status of marketing Authorization
This new treatment for Alzheimer's disease has been conditionally approved by Chinese regulatory authorities, making it China's first self-developed drug for the treatment of the most common form of dementia, which affects about 50 millions of people around the world.
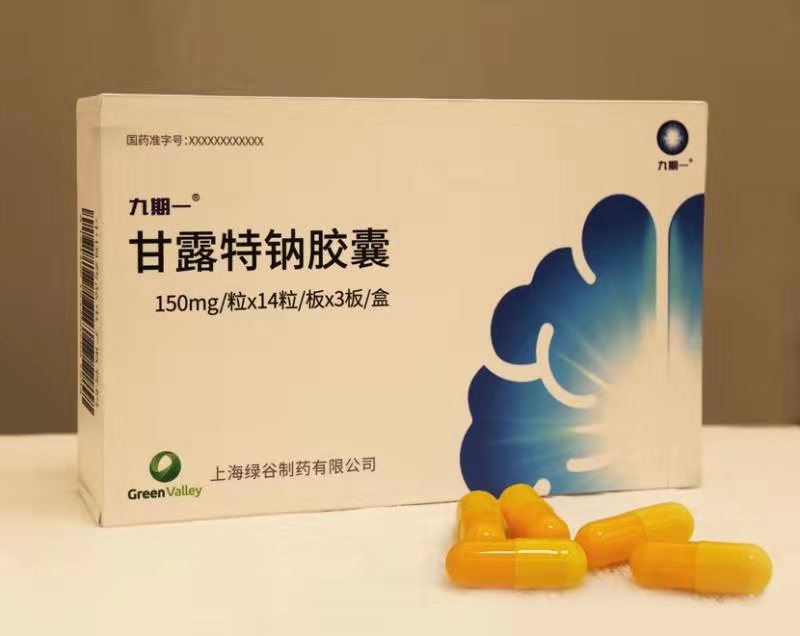
It is a medicine against Alzheimer's disease which derived from marine brown algae.
The drug, approved under China's Priority Review System, was jointly developed by Ocean University of China, Shanghai Medical Materials Institute (SIMM) and Shanghai Green Valley Pharmaceuticals during 22 years of intensive research efforts.
Shanghai Green Valley Pharmaceuticals said its drug oligomannate improved the cognitive function of patients with mild-to-moderate Alzheimer's disease compared to placebo in a phase 3 clinical trial. On November 2, the National Medical Products Administration of China said that oligomannate, is safe for the treatment of "mild to moderate Alzheimer's disease and could improve the cognitive functions of patients," adding that more long-term research is still needed after the approval of the drug.
** GV-971 ** is a mixture of acid linear oligosaccharides ranging from dimer to decamer (molecular weight up to ~ 1 kDa).
How it works
Instead of being designed to eliminate accumulations of protein in the brain, as is the case with dozens of experimental treatments for Alzheimer's disease, oligomannate modulates the relationship between the brain and the bacterial communities of the intestine, which is called microbiome.
For the Chinese researchers, dysbiosis of the gut microbiota involves the infiltration of various peripheral immune cells, including CD4 + and CD8 + T cells, B cells, NK (Natural Killer) cells, neutrophils, dendritic cells (CD ) and monocytes, to the brain. Among them, Th1 cells were particularly noted for their close association with the activation of M1 microglia during the progression of Alzheimer's disease. The authors propose that intestinal dysbiosis promotes infiltration of Th1 cells in order to activate with M1 microglia and trigger its differentiation to a pro-inflammatory state.
Clinical trials
Geng Meiyu, a team scientist and professor at SIMM, told at press conference on Sunday, November 3 that oligomannate was effective in rehabilitating the gut microbiota, describing it as "a great innovation." Lü Songtao, chairman of Green Valley Pharmaceuticals, said the drug will be launched in China by the end of the year and a third phase of clinical trials will be conducted in the United States. Green Valley also announced that it will launch a global Phase 3 trial next year in several other countries. The price of this medicine is still being determined.
The next trial would include more people than the approximately 800 people enrolled in the previous phase 3 trial.
The completed trial was not designed to demonstrate that the drug had apparently functioned according to the mechanism theorized by the researchers, but the next global trial will attempt to show it.
Contact the author
Advertisement

This book retraces the main achievements of ALS research over the last 30 years, presents the drugs under clinical trial, as well as ongoing research on future treatments likely to be able stop the disease in a few years and to provide a complete cure in a decade or two.
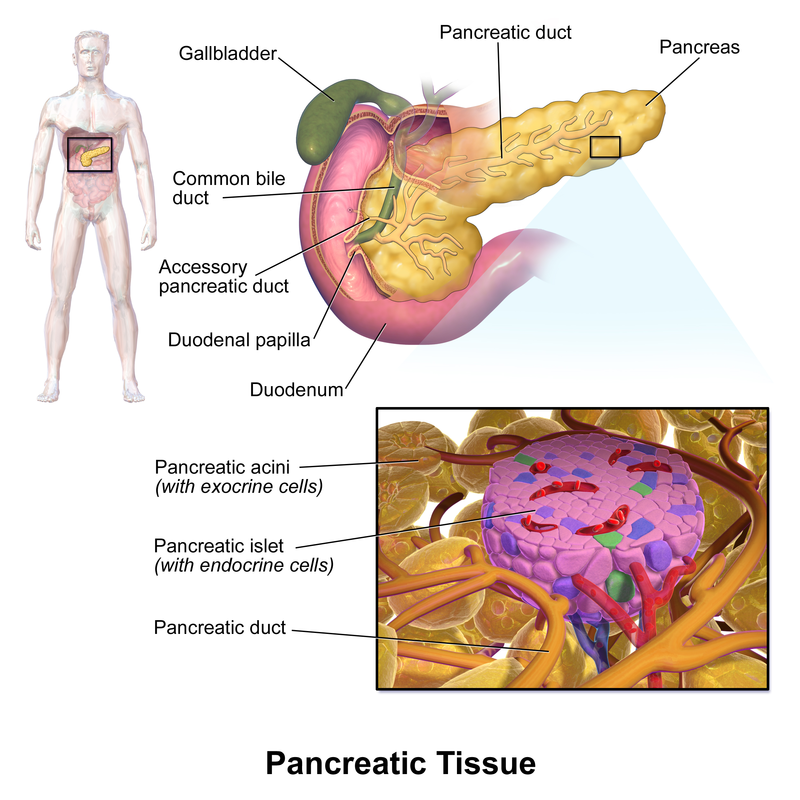 Source: Blausen.com staff (2014). "Medical gallery
Source: Blausen.com staff (2014). "Medical gallery