Inhibiting cGAS-STING pathway confers resilience against Alzheimer's . '
Blood Lipoprotein Levels and Alzheimer Disease . '
Certains médicaments contre le VIH et l’hépatite pourraient contribuer à... . '
Perineuronal net modulation in a Parkinson's disease mouse model . '
Parkinson's disease and acupuncture . '
How to cope with the deluge of scientific publication? . '
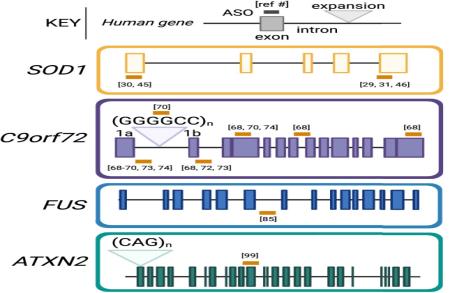
Jacifusen pour FUS-ALS : une étude de cas . '
Small molecule may dissolve stress granules . '
Patient-Derived Cortical Organoids Reveal Senescence of Neural Progenitor Cells in Hutchinson-Gilford Progeria Syndrome. . '
Cerebrovascular variability interactions after acute ischemic stroke: insights from directionality analysis based on... . '
Glucose Metabolism, Lactate, Lactylation and Alzheimer's Disease. . '
Dynamic CTA-Based Whole-Brain Arterial-Venous Collateral Assessment for Predicting Futile Recanalization in Acute... . '
Selective androgen receptor modulators (SARMs) - potential anabolic drugs for the treatment of cachexia and frailty... . '