Inhibiting cGAS-STING pathway confers resilience against Alzheimer's . '
Blood Lipoprotein Levels and Alzheimer Disease . '
Certains médicaments contre le VIH et l’hépatite pourraient contribuer à... . '
Perineuronal net modulation in a Parkinson's disease mouse model . '
Parkinson's disease and acupuncture . '
How to cope with the deluge of scientific publication? . '
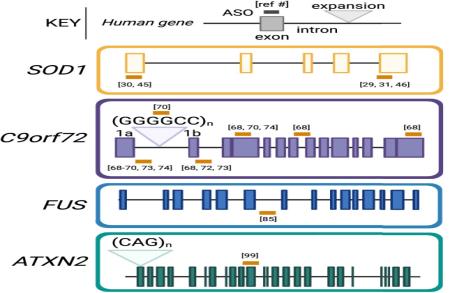
Jacifusen pour FUS-ALS : une étude de cas . '
Small molecule may dissolve stress granules . '
Alternative splicing and the aging brain in AfrAbia: New frontiers in dementia research. . '
An artificial intelligence-derived metabolic network predicts psychosis in Alzheimer's disease. . '
GAN-enhanced deep learning for improved Alzheimer's disease classification and longitudinal brain change analysis. . '
Equivalence of Plasma and Serum for Clinical Measurement of p-tau217: Comparative Analyses of Four Blood-Based Assays. . '
AlzheimerViT: harnessing lightweight vision transformer architecture for proactive Alzheimer's screening. . '