Inhibiting cGAS-STING pathway confers resilience against Alzheimer's . '
Blood Lipoprotein Levels and Alzheimer Disease . '
Certains médicaments contre le VIH et l’hépatite pourraient contribuer à... . '
Does human pegivirus trigger Parkinson's disease? . '
Perineuronal net modulation in a Parkinson's disease mouse model . '
Parkinson's disease and acupuncture . '
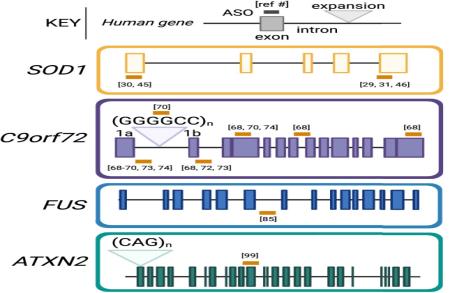
Jacifusen pour FUS-ALS : une étude de cas . '
How to cope with the deluge of scientific publication? . '
Small molecule may dissolve stress granules . '
Epigenetic information loss is a common feature of multiple diseases and aging. . '
Effect of FRAXplus adjustments on fracture risk reclassification in older Swedish women-results from the SUPERB-study. . '
Cell Counting and Cell Cycle Analysis of Simple Non-Cultured Endothelial Cell Injection (SNEC-I) Therapy:... . '
Advances in brain remodeling, stem cell therapies, and translational barriers in stroke and brain aging. . '
Disease Burden of Autism Spectrum Disorder and Attention - Deficit/Hyperactivity Disorder in the 0-14 Age Group across... . '