Alzheimer's disease (AD), a prevalent neurodegenerative disease with progressive dementia in older adults. The dominant hypothesis tells it is caused by abnormal Amyloid-β (Aβ) peptide in extracellular plaques in the brain. A competing hypothesis ponts to the culprit is intracellular Tau aggregates, which would make Alzheimer's disease more similar to other neurodegenerative diseases.
Drugs to cure AD are not in sight, there were more than 2500 unsuccessful clinical trials in Alzheimer's disease.
The authors of a recent article wanted to determine the effect of various neurotransmission-altering compounds including fenobam, quisqualic acid, and dimethyl sulfoxide in the protection against Amyloid-β toxicity.
The well-known C. elegans Alzheimer's disease model, CL4176, in which human Amyloid-β expression is turned on upon a temperature shift to 25 °C that leads to paralysis, was screened for protection/delay in paralysis because of Amyloid-β toxicity.
Still it looks that scientists choose their animal model more because of availability and costs than credibility. Nematode last ancestors in common with Chordate (Protostomia) lived 650 millions years ago, before the Cambian explosion. In contrast last common ancestors between humans and rodents are 60 million years. Nematode's neurons do not fire action potentials, and do not express any voltage-gated sodium channels. A weird choice of animal model for a neurodegenerative disease!
While screening the compounds, dimethyl sulfoxide (DMSO), a universal solvent used to solubilize compounds, was identified to provide protection. DMSO has been examined for the treatment of numerous conditions including dubious alternative medicine "cures".
The scientists conclusion is that DMSO and Fenobam protect against Amyloid-β toxicity through modulation of neurotransmission. Yet this seems far fetched because of the choice of animal model.
 Source: KieranMaher at English Wikibooks
Source: KieranMaher at English Wikibooks
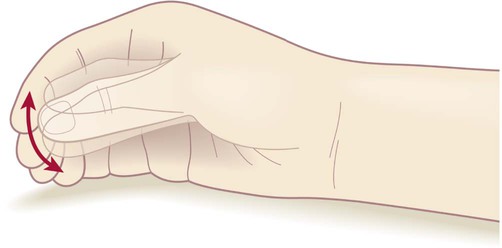 ‘Pill-rolling’ rest tremor as found in Parkinson’s disease.
‘Pill-rolling’ rest tremor as found in Parkinson’s disease.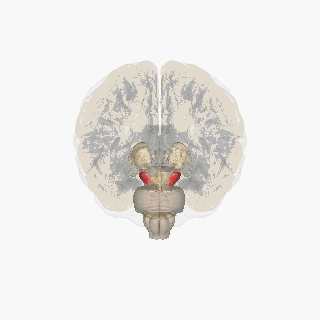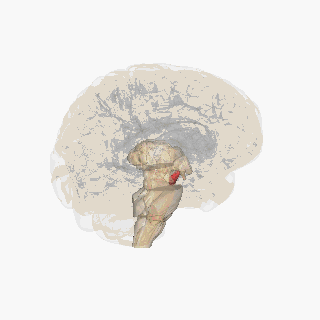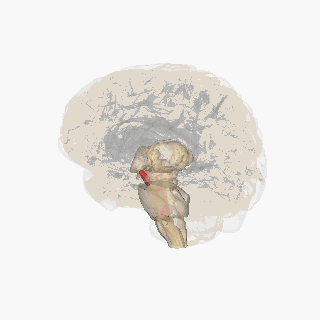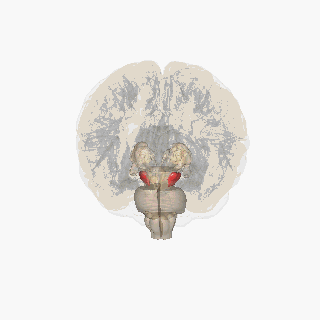 Source Wikipedia: FrozenMan - Own work
Source Wikipedia: FrozenMan - Own work
