Opposing p53 and mTOR/AKT promote an in vivo switch from apoptosis to senescence upon telomere shortening in zebrafish
Mounir El Maï, Marta Marzullo, Inês Pimenta de Castro, Miguel Godinho Ferreira
Voici un article intéressant quand on a en perspective les maladies neuro-dégénérescentes. On y retrouve des termes qui entrent en résonance avec ceux qui nous sont familier : ROS, SOD, résistance à l’insuline. Bien que cet article ne s’applique pas aux neurones (car ils ne se divisent pas) il pourrait fournir une clé pour la SLA et d’autres maladies neurodégénératives.
 Source: https://elifesciences.org/articles/54935/figures
Source: https://elifesciences.org/articles/54935/figures
En effet il présente l'idée que chez un organisme jeune, une cellule défectueuse se suicide et est remplacée facilement, alors que chez un organisme agée une cellule défectueuse cherche à subsister malgré tout, ce qui endommage le tissu où elle est insérée. L'élément déterminant ce changement de comportement étant le contrôle de qualité de l'ADN.
À chaque fois qu’un chromosome d'une cellule eucaryote est répliqué, l'ADN polymérase s'avère incapable de copier les derniers nucléotides. Pour pallier à ce problème, l'ADN d'un chromosome est équipé de télomère à chaque extrémité. C'est une région à l'extrémité d'un chromosome, qui est sans information génétique. Lors de chaque division cellulaire, les télomères s'érodent jusqu'à atteindre une taille critique.
L’arrêt du cycle cellulaire permet à la cellule de stimuler ses mécanismes de réparation, dont certains sont directement activés par la voie p53. Lorsque la réparation est effectuée, le taux de p53 retourne à la normale et le cycle cellulaire reprend. Si les dommages subis par la cellule ne peuvent pas être réparés, la cellule entre en apoptose, ce qui conduit à son élimination.
Une prolifération cellulaire est cependant possible par inactivation de la voie p53. Les cellules entrant dans la prolifération après l'inactivation de la voie p53 subissent des réarrangements chromosomiques grossiers et présentent une instabilité de leur génome.
Dans cet article, les auteurs étudient l’effet de la réduction des télomères chez un animal modèle (poisson zèbre) où cette réduction est accélérée.
Chez la plupart des eucaryotes, le raccourcissement des télomères est contrecarré par la télomérase, bien que son expression soit limitée dans la plupart des cellules somatiques humaines. Par conséquent, les télomères raccourcissent considérablement pendant le vieillissement humain. Mais, et ce n’est pas explicité dans l’article, cela n’est vrai que pour les cellules qui sont l’objet d’un remplacement fréquent.
Curieusement alors que les maladies neurodégénérescentes sont liées à l’âge, les neurones ne sont pas soumis à la limite de Hayflick, même s’il y en a une pour les cellules gliales. Peut-être que le mécanisme décrit dans cet article, induit des maladies neurodégénérescentes par son action sur les cellules gliales?.
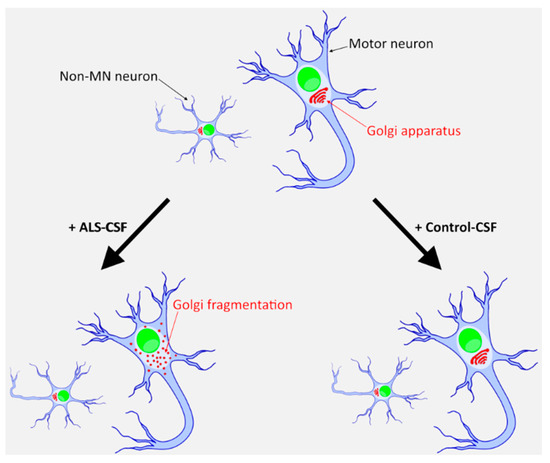
On peut se demander dans quelle mesure la sénescence des neurones et autres cellules gliales, entraîne des protéopathies. Par exemple s’il n’y a pas assez d’énergie, le réticulum endoplasmique ne pourra pas assurer son rôle de conformation des protéines, et pareillement l’appareil de Golgi ne pourra délivrer ces protéines à leur destination normale. Cela pourrait entraîner une protéopathie.
 Source: Magnus Manske via Wikipedia
Source: Magnus Manske via Wikipedia
Mais ce qui motive cet article c’est que le mécanisme moléculaire déterminant le destin cellulaire (apoptose ou sénescence) reste incertain.
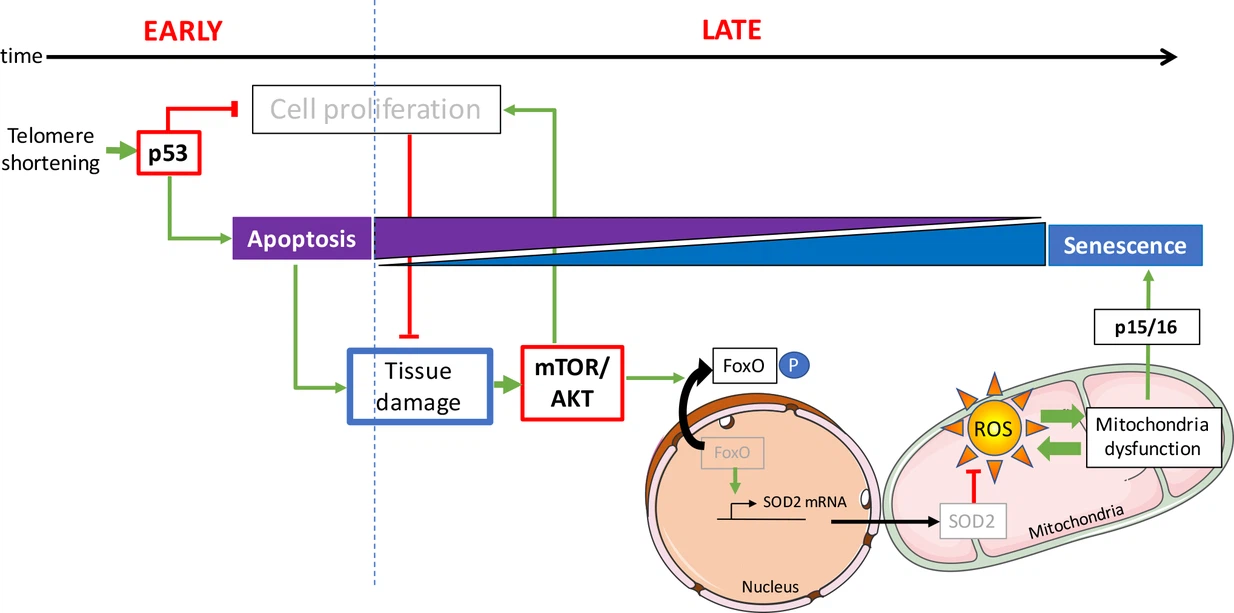
Dans la présente étude, les auteurs décrivent des poissons zèbres jeunes (âgés de 3 mois) , qui sont déficients en télomérase présentent déjà des dommages à l‘ADN et une activation de p53. A ce stade, l’apoptose est le destin cellulaire prédominant. Même si des dommages à l’ADN sont présents dans les tissus prolifératifs, comme l’intestin et les testicules, aucun signe de sénescence cellulaire n’a pu être détecté. Au cours de cette étude, l’activation précoce de p53 chez le poisson zèbre ne modifie pas la fonction des mitochondries.
Cependant, il existe un changement évident entre l’apoptose et la sénescence chez les poissons plus âgés. Chez ces animaux, la sénescence devient la réponse cellulaire la plus répandue. Cette observation souligne le fait que les mêmes types de cellules peuvent subir des destins cellulaires différents in vivo en fonction de l’âge de l’animal.
Au contraire des poissons jeunes, l’intestin et les testicules des poissons zèbres plus âgés présentent un dysfonctionnement mitochondrial accompagné d’une réduction significative des niveaux d’ATP (l'ATP fournit l'énergie nécessaire aux réactions chimiques du métabolisme cellulaire) et d'une accumulation de ROS (bien qu'il s'agisse de sous-produits du métabolisme normal de l'oxygène, leur concentration peut croître significativement en période de stress et endommager les structures cellulaires).
L’étude révèle que les défauts mitochondriaux sont associés à une réduction des défenses mitochondriales. Les auteurs observent qu’avec l’âge, l’expression de SOD2 est réduite en réponse à l’activation de la voie AKT.
Chez l'homme, il existe 3 gènes de la famille Akt : Akt1, Akt2, et Akt3. Ces enzymes appartiennent à la famille des protéines kinases. La protéine kinase B (PKB), également connue sous le nom d'Akt, est une protéine kinase spécifique à la sérine/thréonine qui joue un rôle clé dans plusieurs processus cellulaires tels que le métabolisme du glucose, l'apoptose, la prolifération cellulaire, la transcription et la migration cellulaire. L'AKT est activée sur des signaux extracellulaires pro-prolifératifs. La voie mTOR / AKT est déclenchée par les récepteurs du facteur de croissance, y compris le récepteur du facteur de croissance de l'insuline (IGFR).
Les voies anti-proliférative p53 et les voies pro-survie mTOR/AKT interagissent de manière complexe. Selon le contexte, l’interaction de ces voies module le destin cellulaire en arrêt du cycle cellulaire, apoptose ou sénescence. Conformément à ces données in vitro, les scientifiques montrent que l’activation de l’AKT chez le poisson zèbre mutant âgé est concomitante avec une suppression de l’apoptose et un l’activation de mécanismes entraînant la sénescence.
Quelle est le mécanisme qui fait basculer le destin cellulaire de l’apoptose à la sénescence? Un taux d'apoptose élevée parmi les cellules d'un tissu, augmente la demande de prolifération cellulaire des cellules environnantes dans un processus appelé prolifération compensatoire induite par l’apoptose.
Au contraire les restrictions de prolifération cellulaire entraînent une dégénérescence tissulaire qui devient apparente chez les poissons zèbres vieillissants.
Dans les tissus où les cellules souches ne sont pas facilement disponibles ou lorsque les programmes génétiques intrinsèques aux tissus limitent la division cellulaire, l’hypertrophie cellulaire représente une stratégie alternative à l’homéostasie tissulaire.
Publicité

Ce livre retrace les principales réalisations de la recherche sur la SLA au cours des 30 dernières années. Il présente les médicaments en cours d’essai clinique ainsi que les recherches en cours sur les futurs traitements susceptibles d’ici quelques années, d’arrêter la maladie et de fournir un traitement complet en une décennie ou deux.