Parkinson's disease is characterized by a loss of dopaminergic neurons in the substantia nigra. There is no treatment that can improve the course of Parkinson's disease. While most treatment strategies are aimed at preventing neuronal loss or protecting neural circuits, a potential alternative, is to replace lost neurons to reconstruct altered neural circuits.
Given the plasticity of some somatic cells, transdifferentiation approaches to change the fate of cells (in situ as escape the immune system) have gained momentum. In the brain of mice, the plasticity of glial cells has thus been used to generate new neurons which have shown an improvement in disease in model animals.
Most in vivo reprogramming relies on the use of transcription factors specific to the cell line under consideration. This study shows that there are other ways to achieve this goal.
Researchers from the University of California (UC), San Diego School of Medicine, have developed a non-infectious virus that carries an antisense oligonucleotide sequence designed to specifically bind to RNA encoding the PTB protein, degrading it, preventing it from being translated into a functional protein. Antisense oligonucleotides are a proven therapeutic approach.
Sequential downregulation of PTB and nPTB occurs naturally during neurogenesis, and once triggered, the gene expression loops regulated by PTB and nPTB become self-reinforcing. By modulating the two loops, the sequential negative regulation of PTB and nPTB makes it possible to generate functional neurons from human fibroblasts.
Astrocytes offer several benefits for in vivo reprogramming in the brain. These non-neuronal cells are abundant, proliferate when injured and are very plastic. They can adopt different phenotypes or even be reprogrammed in a very different cell type. Astrocytes can be converted into different neural subtypes, depending on their region of origin in the brain.
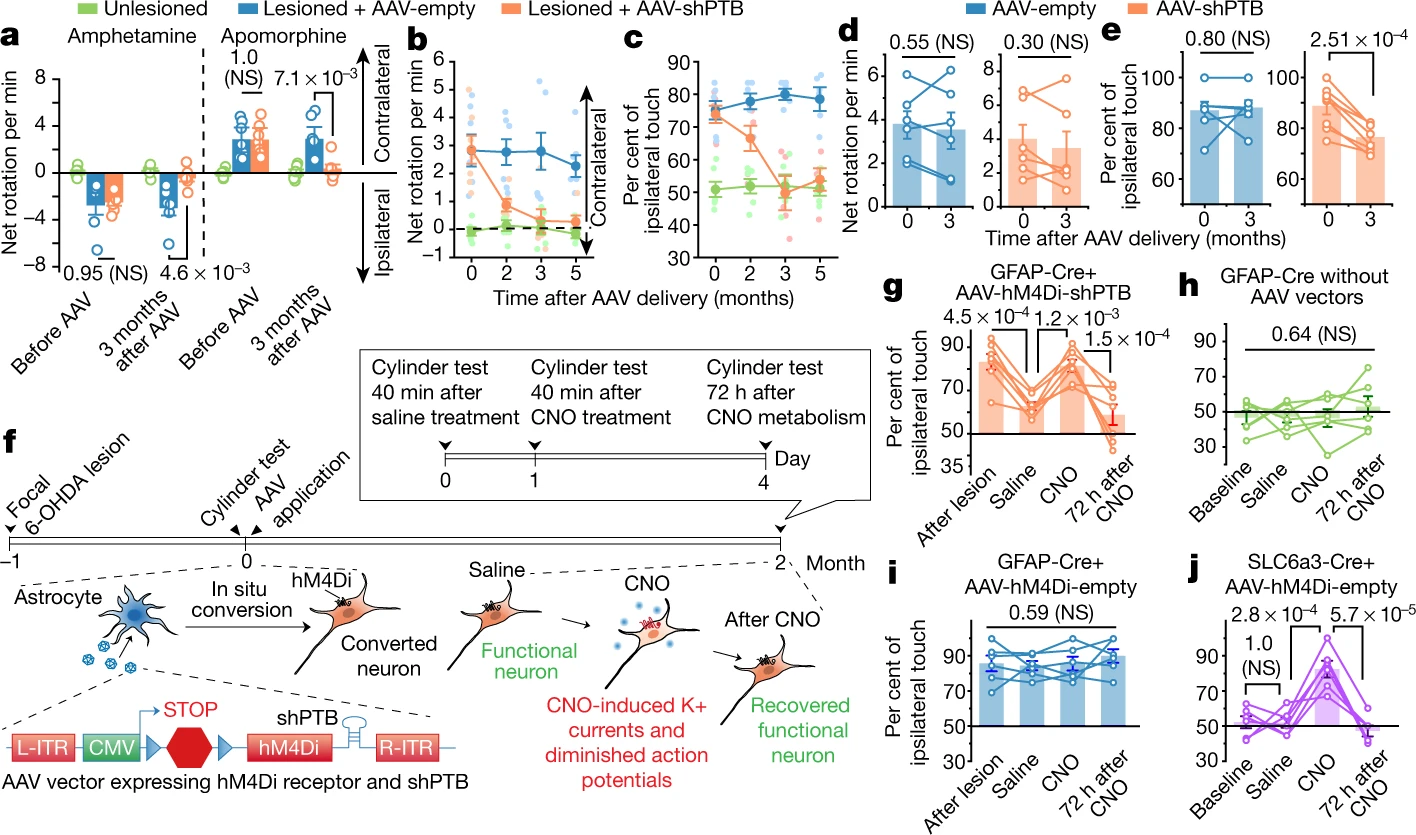
Here, scientists from the University of California, San Diego School of Medicine, report an efficient, single-step conversion of astrocytes from humans and mice into functional neurons. This by depleting the RNA-binding protein PTB (also known as PTBP1).
The target cells for this conversion are the dopaminergic neurons in the substantia nigra, that is, those that become non-functional in Parkinson's disease. Using this approach, the team demonstrated the gradual conversion of astrocytes into new neurons capable of innervating and repopulating the neural circuits of the substantia nigra. These dopaminergic neurons induced by depletion of the PTB powerfully restore striatal dopamine, restore the nigrostriatal circuit and reverse the motor phenotypes of Parkinson's disease.

In treated mice, a subset of about 30% of the astrocytes were converted to neurons, thus increasing the total number of neurons. Dopamine levels were restored to a level comparable to that of normal mice. In addition, neurons have developed and sent their processes to other parts of the brain. There was no change in the control mice.
The treated mice recovered their vitality with a single treatment and remained completely free of Parkinson's symptoms for the rest of their lives. In contrast, control mice did not show any improvement.
To experiment with the conversion of midbrain astrocytes to dopaminergic neurons, the scientists used a model of chemically induced Parkinson's disease in mice. The model used by the team does not perfectly summarize all the essential characteristics of Parkinson's disease. In the future, scientists will use a more expensive animal genetic model of Parkinson's.
One could wonder if this therapy can be transposed to other neurodegenerative diseases. However, Parkinson's disease is characterized by an illness in a very specific region of the brain. On the contrary, in Alzheimer's disease, the damage is global to the brain and in the case of amyotrophic lateral sclerosis the neurons involved are the motor neurons, but they cover a considerable area.