Alors que les astrocytes aident normalement à lutter contre le dépôt de plaques de β-amyloïdes, une réactivité soutenue des astrocytes pourrait entraîner une dystrophie des astrocytes, l'atrophie de la substance grise et l'hypométabolisme du glucose.

Dans cette nouvelle étude, les scientifiques Nicholas R Livingston, Paul Edison et leurs collègues de l'Imperial College, Londre, ont utilisé le nouveau traceur TEP du récepteur d'imidazoline 11C-BU99008 pour tester l'hypothèse d'une relation dynamique entre la réactivité des astrocytes et la neurodégénérescence.
Ils ont trouvé des indices d'une réactivité accrue des astrocytes chez les patients ayant un développement pathologique des plaques amyloïdes, principalement dans les régions frontale, pariétale et occipitale.
Ces augmentations étaient plus importantes chez les patients atteints de troubles cognitifs légers que chez les patients atteints de la maladie d'Alzheimer.
Des analyses complémentaires ont montré que la réactivité plus faible des astrocytes chez les patients avec un développement pathologique des plaques amyloïdes était associée à la fois à un hypométabolisme du glucose dans les lobes pariétal, temporal et frontal et à une atrophie de la substance grise dans les lobes frontaux et temporaux.
Cependant, un plus grand dépôt de plaques de β-amyloïdes était associé à une réactivité accrue des astrocytes dans les zones corticales primaires motrices et sensorielles primaires, mais une réactivité des astrocytes diminuée dans les régions temporales.
11C-BU99008 est un nouveau traceur PET qui se lie à I2-BS, dont l'expression est associée à la réactivité des astrocytes.
Il existe trois classes principales de récepteurs d'imidazoline : I1 est impliqué dans l'inhibition du système nerveux sympathique pour abaisser la tension artérielle, I2 a des fonctions encore incertaines mais est impliqué dans plusieurs troubles psychiatriques, et I3 régule sécrétion d'insuline. La protéine I2-BS localisée dans le cerveau, est régulé positivement avec le vieillissement en bonne santé, et est encore augmenté dans la maladie d'Alzheimer.
Différentes études passées ont renforcé l'hypothèse selon laquelle la réactivité des astrocytes est un événement précoce dans la progression de la pathologie de la maladie d'Alzheimer, se produisant en réponse à un dépôt amyloïde précoce, qui prend généralement naissance dans le lobe frontal.
Dans les premiers stades, les astrocytes réactifs ont un rôle neuroprotecteur, aidant à la clairance de plaques de β-amyloïdes.
Dans la cohorte étudiée par les scientifiques, ils ont découvert une liaison plus faible du 11C-BU99008 dans le lobe temporal, qui était associée à une progression relative plus importante de la neuropathologie associée à l'amyloïde, c'est-à-dire l'hypométabolisme du glucose et l'atrophie de la matière grise.
Les scientifiques proposent que cette absorption réduite de 11C-BU99008 dans la région du lobe temporal reflète une dystrophie des astrocytes, provoquée par un phénotype d'astrocytes chronique pro-inflammatoire et neurotoxique induit par l'amyloïde et entraînant une capacité glycolytique réduite et une altération secondaire du métabolisme neuronal ou une perte cellulaire.
Il y a des limites évidentes à l'étude des scientifiques.
Premièrement, seul un petit nombre de sujets a pu être examiné.
Une deuxième limitation était la conception transversale, que les scientifiques reconnaissent ; cependant, la pathologie post mortem a la même limitation.
Les résultats des auteurs sont donc mieux interprétés de manière descriptive et suggérant un modèle hypothétique, plutôt qu'un test solide et indépendant.
Néanmoins, la cohérence des directions d'effet observées dans cette étude et les études TEP 11C-DED antérieures fournit un soutien convaincant pour le modèle propos, où la réactivité des astrocytes se produit en réponse à un dépôt précoce de plaques de β-amyloïdes, aidant à la clairance de plaques de β-amyloïdes, mais où après un certain temps les astrocytes deviennent neurotoxiques, contribuant à une activité tissulaire réduite et à la mort cellulaire associée à une déficience cognitive.
En conclusion, les scientifiques ont démontré in vivo avec le nouveau traceur TEP 11C-BU99008 que la réactivité des astrocytes est augmentée dans les régions supposées représenter les premiers stades de la progression pathologique avec de faibles charges de dépôt de plaques de β-amyloïdes, et inversement relativement réduite dans les régions qui montrent des signes de progression plus avancée de la maladie avec un plus grand dépôt de plaques de β-amyloïdes et une atrophie.
Advertisement

This book retraces the main achievements of ALS research over the last 30 years, presents the drugs under clinical trial, as well as ongoing research on future treatments likely to be able stop the disease in a few years and to provide a complete cure in a decade or two.
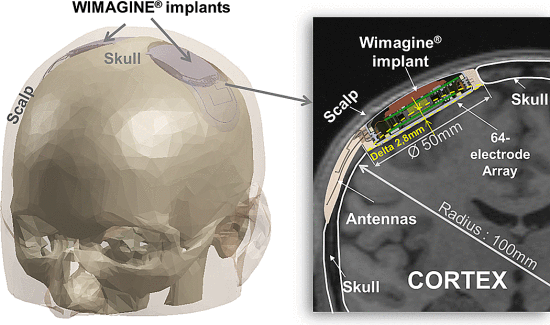
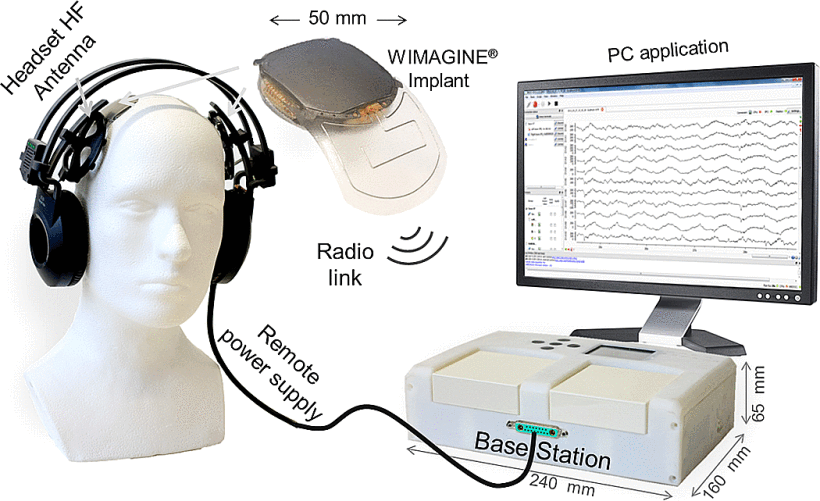
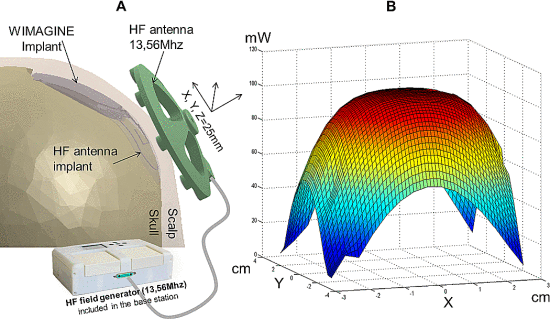
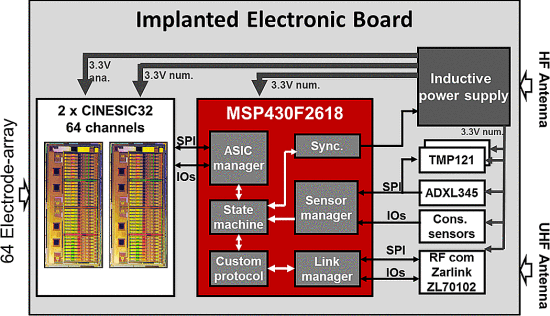
 L 型结构由弱相互作用保持,最终完全错误折叠和寡聚化,形成富含 β-折叠的“β 型”。 L ⇌ β 转换期间蛋白质获得结构的非结构化区域。
L 型结构由弱相互作用保持,最终完全错误折叠和寡聚化,形成富含 β-折叠的“β 型”。 L ⇌ β 转换期间蛋白质获得结构的非结构化区域。 几项观察性队列和病例对照研究表明,接种某些类型的疫苗后痴呆症发生率降低。
二十年前,Verreault 和他的同事报告说,疫苗暴露(白喉/破伤风、脊髓灰质炎、流感)与随后阿尔茨海默病的发展减少 25-60% 相关。
几项观察性队列和病例对照研究表明,接种某些类型的疫苗后痴呆症发生率降低。
二十年前,Verreault 和他的同事报告说,疫苗暴露(白喉/破伤风、脊髓灰质炎、流感)与随后阿尔茨海默病的发展减少 25-60% 相关。 资料来源:Ajpolino 通过维基百科
资料来源:Ajpolino 通过维基百科