This blog mainly deals with the three main neurodegenerative diseases, Alzheimer's, Parkinson's and ALS. Unfortunately, in real life, neurodegenerative diseases do not have textbook like, well-differentiated symptoms.
When Parkinson's and ALS are often associated with dementia, in scientific literature it is less common to associate Parkinson's and Autism.

A recent prepublication article on MedRxiv, written by Kadalraja Raghavan, Samuel JK Abraham and colleagues from India and Japan, indicates that a diet derived from a black fungus may have alleviated the symptoms of autism in children with severe symptoms, and even more surprisingly, it may have lowered their level of α-synuclein in their blood.
No mechanism, intervention or therapy has proven its ability to regulate α-synuclein levels. In this study, plasma α-synuclein levels showed a significant increase after Nichi Glucan supplementation.
Parkinson's disease is a progressive neurodegenerative disease characterized by resting tremors, rigidity, postural instability, and bradykinesia.
Parkinson's disease can also present a wide range of non-motor symptoms, such as autonomic dysfunction, cognitive impairment, sleep disturbances, and neuropsychiatric symptoms, including depression, anxiety, and repetitive or obsessive-compulsive behaviors.
Parkinson's disease is characterized by intracellular inclusions comprising dozens of proteins but mainly α-synuclein. The dopaminergic neurons with their long disproportionate axons that are projecting from substantia nigra (SN) into the striatum are very vulnerable to those protein aggregates.
Autism spectrum disorders are characterized by disorders of social interaction and repetitive and stereotypical behaviors. Additional features that may accompany autism spectrum disorders are motor abnormalities, gastrointestinal issues, epilepsy, intellectual disability, or sleep disturbances.
Although α-synuclein is an intracellular protein predominantly located in the brain, several studies have reported its presence in plasma as well as in cerebrospinal fluid (CSF). The levels of α-synuclein are known to be significantly lower in patients with autism spectrum disorders than in healthy controls.
Measuring α-synuclein in CSF would give more accurate results, but this procedure is invasive and is not ideal for routine monitoring.
In this new, prospective, open-label, two-arm pilot clinical trial, six children with ASD received conventional treatment including corrective behavioral therapy and L-carnosine 500 mg per day, and 12 children received a supplementation of 0,5 g of Nichi Glucan twice a day in addition to the conventional treatment. There was only one female subject in both groups. The study lasted 90 days. Only one child had possible mild side effects related to increased stools for a week.
Subjects included in the study had been diagnosed with ASD by a developmental pediatrician, a diagnosis verified by a psychologist using a clinical interview for a behavioral model that incorporated the Childhood Autism Rating Scale (CARS).
The four children in the control group were in the severe autism category and their mean baseline score was 42.75 ± 5.76. Of the nine children who consumed Nicho glucan, two of them had mild to moderate autism (mean = 33.5 ± 2.5), while the remaining seven were in the severe autism category (mean = 43.71 ± 4.80). After the intervention, the mean CARS score in the four children in the control group was virtually unchanged at 42.5 ± 5.4.
In the Nichi Glucan group, the mean CARS score in the two children with mild to moderate autism was unchanged at 32.5 ± 0.5, while in the other seven children the CARS score improved. slightly (mean = 40.1 ± 5.96).
In the latter group, there was a visible subjective reduction in irritability and anger, improvement in sleep, speech characteristics, and improved caregiver responses.
α-Synuclein has recently been considered one of the important biomarkers for the diagnosis of autism and ASD, where levels are low compared to controls of the same age (Kadak et al., 2015; Sriwimol et al. ., 2018; Siddique et al. 2020). Plasma α-synuclein levels were significantly higher in the Nichi Glucan group than in the control group.
After the operation, plasma α-synuclein levels increased on average by 26.72 ng/dl. Human α-synuclein plasma levels were measured in peripheral blood at baseline and at the end of the 90-day study.
Β-glucans are polysaccharides naturally present in the cell walls of cereals, bacteria and fungi. The β-glucans found in yeast cell walls contain a 1.3 carbon backbone with elongated 1.6 carbon branches. Nichi Glucan, is a food supplement containing β-1,3-1,6-Glucan produced by black yeast (strain AFO-202).
At dietary intake levels of at least 3 g per day, the β-glucan in oat fiber lowers blood LDL cholesterol levels and thus may reduce the risk of cardiovascular disease. Β-Glucans are also used in various nutraceuticals and cosmetics.
It should be noted that Dr. Abraham is a shareholder in GN Corporation, Japan which in turn is a shareholder in the manufacturing company of the AFO 202 Beta Glucan

 How ASOs work in the human body. Image by Larissa Nitschke
How ASOs work in the human body. Image by Larissa Nitschke
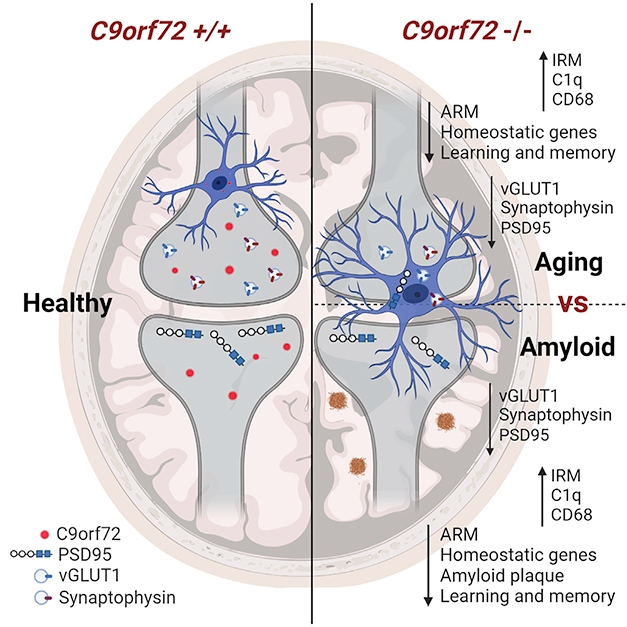

 Cellular stress response is an umbrella concept for molecular changes that cells undergo in response to exposure to extreme temperature, viral infection, toxins, stroke or injury.
Cellular stress response is an umbrella concept for molecular changes that cells undergo in response to exposure to extreme temperature, viral infection, toxins, stroke or injury. 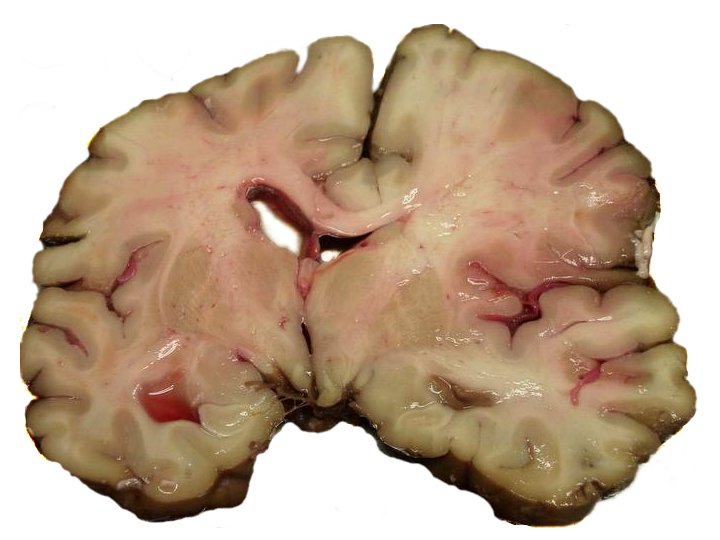 A slice of brain of a person who had a stroke
By Marvin 101 - via Wikipedia
A slice of brain of a person who had a stroke
By Marvin 101 - via Wikipedia