Despite often being described as two separate neurodegenerative disorders, several independent studies confirmed that clinical symptoms and pathologies of Alzheimer's disease and Parkinson's disease can overlap.
Indeed dementia with Lewy bodies involves deposition of both extra-cellular amyloid-β plaques and intra-cellular α-synuclein containing Lewy bodies which are pathological hallmarks of Alzheimer's disease and Parkinson's disease respectively.
 Robin Williams (pictured in 2011) was diagnosed during autopsy as having diffuse Lewy bodies.
Robin Williams (pictured in 2011) was diagnosed during autopsy as having diffuse Lewy bodies.
Scientists correlated two interesting observations: - First, humans as well as mice with a strong α-synuclein pathology, appear to have a lower burden of amyloid-β. - Second, a common feature of neurodegenerative diseases is the occurrence of olfactory dysfunctions at a very early stage of disease progression, even years before other clinical symptoms occur
Diagnosis of Dementia with Lewy bodies is extremely difficult and often leads to a misdiagnosis of Alzheimer's disease or Parkinson disease. Until now, an accurate diagnosis can only be made by postmortem analysis. So there is an urgent need of biomarkers to improve identification of dementia subtypes.
Therefore, studies of early pathological alterations in the olfactory bulb are of great interest in order to use olfactory deficits as a biomarker for early diagnoses and disease progression.
In the present study, the scientists therefore examined the effect of α-synuclein on the formation of amyloid-β aggregates specifically in the olfactory bulb.
They shown on a mice model of dementia with Lewy bodies, that as α-synuclein increases, the amyloid-β load in the olfactory bulb of these mice diminishes and that led to a significant amelioration of olfactory performance.
Via inoculation of amyloid-β rich brain extracts in the olfactory bulb, the authors could show that the amyloid-β seeding area was significantly decreased in the olfactory bulb of aged (8 months) mice model of dementia with Lewy bodies compared to same age mice with a model of Alzheimer's disease.
Experiments with grafts of embryonic neurons supported the inhibitory role of α-synuclein as the number of plaques was significantly reduced when healthy grafts were transplanted into a mouse model of Lewy body dementia, compared to the same graft in a mice model of Alzheimer's disease.
The scientists determined that the decreased Amyloid-β plaque load in 8-month-old mice was not attributable to an increased phagocytic capacity of microglial cells. Extra-cellular α-synuclein seems to interfere with the formation of Aβ deposits.
In conclusion, this study verifies an inhibitory role of α-synuclein on amyloid-β plaque formation in the olfactory bulb of a mice model od Dementia with Lewy body mice, which further lead to the amelioration of olfactory deficits in mice harbouring both α-synuclein and amyloid-β pathologies.
Indeed this work was done on mice models of diseases, and it rarely translate in similar human findings. In addition it is not clear if these mice were standardized commercial products or in-site raised animals. Finally the authors do no explain what is the relation between the extra-cellular α-synuclein, which was the subject of their study and the intra-cellular α-synuclein found in Lewy bodies.
So anosmia (loss of the ability to detect one or more smells) might be a clinical sign helping to differentiate Alzheimer disease from Dementia with Lewy bodies

 Source: KieranMaher at English Wikibooks
Source: KieranMaher at English Wikibooks
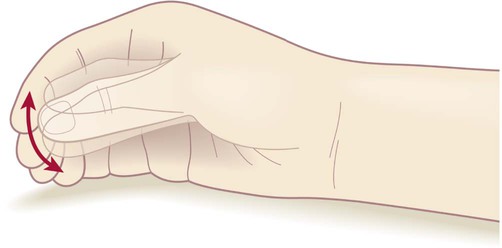 ‘Pill-rolling’ rest tremor as found in Parkinson’s disease.
‘Pill-rolling’ rest tremor as found in Parkinson’s disease.
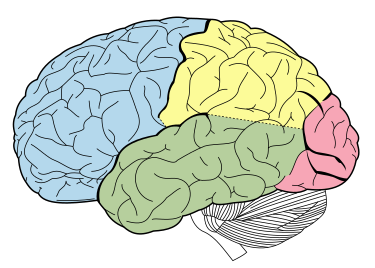 Source: Wikipedia. The temporal lobe is shown in green, while the motor area is in the frontal lobe in blue
Source: Wikipedia. The temporal lobe is shown in green, while the motor area is in the frontal lobe in blue Hagmann P, Cammoun L, Gigandet X, Meuli R, Honey CJ, et al.
Hagmann P, Cammoun L, Gigandet X, Meuli R, Honey CJ, et al.  In silico computer models for DBS allow to pre-select a set of potentially optimal stimulation parameters. If efficacious, they could further carry insight into the mechanism of action of DBS and foster the development of more efficient stimulation approaches. In recent years, the focus has shifted towards DBS-induced firing in myelinated axons, deemed particularly relevant for the external modulation of neural activity.
In silico computer models for DBS allow to pre-select a set of potentially optimal stimulation parameters. If efficacious, they could further carry insight into the mechanism of action of DBS and foster the development of more efficient stimulation approaches. In recent years, the focus has shifted towards DBS-induced firing in myelinated axons, deemed particularly relevant for the external modulation of neural activity.