A new publication presents work on the link between a protein called IL-11 and aging in mice.
These studies identify IL11 as a key inflammatory factor and therapeutic target for mammalian health. What is new is that this effect is positive even for elderly subjects and for both sexes.
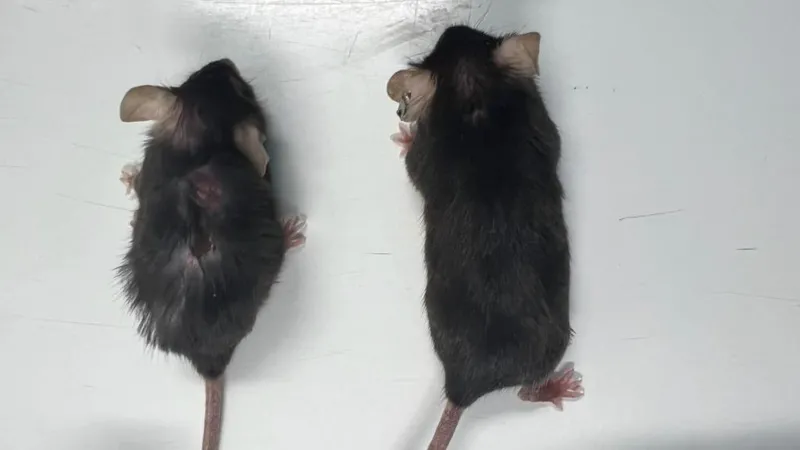 IL11 belongs to the IL6 family of cytokines, which bind to alpha receptors and the gp130 coreceptor to initiate intracellular signaling via JAK/STAT. This family is evolutionarily ancient, with homologs found in ascidians and fish.
IL11 belongs to the IL6 family of cytokines, which bind to alpha receptors and the gp130 coreceptor to initiate intracellular signaling via JAK/STAT. This family is evolutionarily ancient, with homologs found in ascidians and fish.
The authors found that IL-11 levels increase with age and that inhibition of IL-11 provides several benefits, including improved metabolism, muscle function, and duration of life. Inhibition also appears to reduce age-related cancers.
Researchers believe that inhibiting IL-11 could be a promising approach to extending human lifespan and health, especially since drugs targeting IL-11 are already being tested for safety in the framework of clinical trials.
Initially perceived as anti-fibrotic, anti-inflammatory, and pro-regenerative, IL11 could be pro-fibrotic, pro-inflammatory, and anti-regenerative. Two misinterpretations of previous studies have shaped this incorrect understanding.
Identified in 1990 as a factor secreted by bone marrow cells, IL11 appeared to be synergistic with IL3 for the formation of megakaryocytes and the increase in platelets. This belief was validated by studies using recombinant human IL11 (rhIL11), leading to its approval by the FDA in 1998 to treat thrombocytopenia. However, as early as 1997, studies suggested that IL11 had primary functions other than hematopoiesis.
The putative role of IL11 as a hematopoietic factor has led to the development of rhIL11 as an antifibrotic, anti-inflammatory, and pro-regenerative in murine models. Clinical trials have been carried out on various diseases, but without conclusive success, suggesting a lack of effectiveness or significant toxicities.
In 2016, IL11 was shown to activate fibrogenic proteins in human fibroblasts, causing a disease: Fibrosis. This finding contradicted 20 years of data showing beneficial effects. rhIL11 appeared to inhibit the function of endogenous IL11 in mice, explaining the beneficial effects observed in previous studies. The authors of these studies did not understand that the observed effects reflected rhIL11-mediated inhibition of endogenous IL11.
From the beginning of IL11 studies, it was observed that IL11 is upregulated in tissues of aged rodents. This discovery spurred a six-year (2017-2023) study of IL11 in terms of lifespan and health. During this period, it became clear that IL11 is part of the senescence-associated secretory phenotype and can directly stimulate senescence in lung fibroblasts and epithelial cells.
Serum levels of IL11 are increased in very elderly people. Healthspan studies have identified IL11 as an inflammatory factor responsible for ERK/mTOR-mediated sarcopenia, metabolic dysfunction, and frailty in aged mice while showing that IL11 inhibition increases the mouse's health duration.
Chronic inflammation is an important feature of aging, intimately linked to senescence and implicated in the pathogenesis of age-related frailty, metabolic dysfunction, and multimorbidity. Studies in invertebrates have shown that innate immune signaling, including Jak-Stat signaling in fly adipose tissue, can impair metabolism and lifespan. The relative contributions of canonical (JAK–STAT3) and noncanonical (MEK–ERK) IL-11 signaling, alone or in combination, to aging phenotypes remain to be determined.
Inhibition of ERK or mTOR or activation of AMPK by trametinib, rapamycin, or metformin, respectively, increases lifespan in model organisms and some advocate the use of these drugs in humans. However, sometimes these agents have detrimental, effects on health and inflammation.
As mice age, IL11 gradually appears in the liver, skeletal muscle, and fat. to stimulate an ERK/AMPK/mTORC1 axis of cellular, tissue, and organism aging pathologies. In aged mice, the deletion of Il11 protects against metabolic multimorbidity, sarcopenia, and frailty. Administering anti-IL11 treatment to aged mice for six months reactivates an age-repressed white fat loss program, reverses metabolic dysfunction, restores muscle function, and reduces frailty. In all cases studied, IL11 inhibition lowers epigenetic age, reduces telomere attrition, and preserves mitochondrial function.
The metabolic effects observed with IL-11 inhibition in aged mice resemble those of young mice with white adipose-specific Raptor deletion. White adipose tissue (fat) and brown adipose tissue are the two main types of adipose tissue in mammals. The authors speculate that inhibition of IL-11 prevents mTORC1 activation in fat, thereby decreasing the amount of white adipose tissue. However, the authors did not identify the mechanism leading to weight loss with IL-11 inhibition, which in further studies could inform some of the present findings.
Beyond metabolism, IL-11 inhibition ameliorated deterministic features of aging common in vertebrates (such as frailty and sarcopenia), demonstrating generic anti-aging benefits at the organismal level. Intriguingly, some of the beneficial effects of germline Il11ra1 or Il11 deletion, notably on muscle and fat, were apparent even in young mice. The authors did not see any specificity of this effect to certain tissues.
Inhibition of IL-11 increased lifespan in both male and female mice. The extent of the increase in lifespan remains unclear, but current data suggest that anti-IL-11 treatment given late in life increases median lifespan by more than 20% in both sexes. In these experiments, anti-IL-11 was injected into mice at 75 weeks (human equivalent to approximately 55 years).
Treatment with IL-11 is therefore effective in prolonging lifespan, as is the case for rapamycin. The mortality of mice in the elderly is often linked to cancer and the end-of-life autopsy data carried out by the authors support the idea that inhibition of IL-11 considerably reduces cancers linked to 'age. Of note, IL-11 is often linked to tumor onset and immune evasion. Clinical trials of anti-IL-11 in combination with immunotherapy to treat cancer are already planned.
As a first step toward creating a drug, the authors designed a high-affinity humanized neutralizing IL11 antibody.
It should be noted that some of the authors have strong links with companies that have acquired the patents resulting from this work or financed this work.
Also, the green and yellow vegetables and fruits, such as leafy greens, herbs, broccoli, peas, green bell peppers, and squash, are relatively rich sources of the oxycarotenoids lutein and zeaxanthin which may prevent the overexpression of IL-11 and ERK signaling
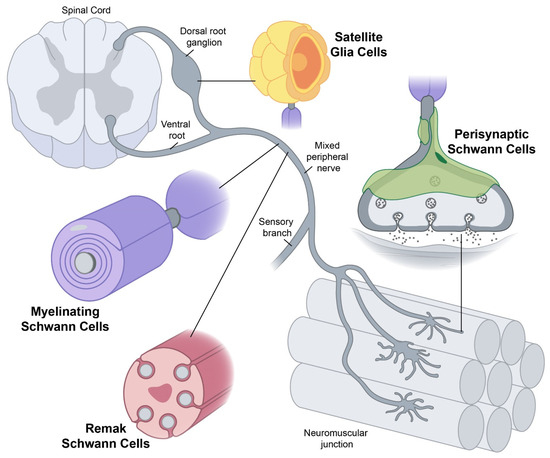 Yet conceptually associating Schwann cells and ALS is not common, ALS is a disease of the central nervous system (upper motor neurons in the brain and spine) while Schwann cells are located in the peripheral nervous system (lower motor neurons with their bodies from the spine and terminating in muscles). This does not mean there are no relations between the two types of cells, and it's a common view now that neurons are not independent, self-sufficient entities and that they are cared for by a large number of other cell types.
Yet conceptually associating Schwann cells and ALS is not common, ALS is a disease of the central nervous system (upper motor neurons in the brain and spine) while Schwann cells are located in the peripheral nervous system (lower motor neurons with their bodies from the spine and terminating in muscles). This does not mean there are no relations between the two types of cells, and it's a common view now that neurons are not independent, self-sufficient entities and that they are cared for by a large number of other cell types. Telomerase reverse transcriptase (abbreviated to TERT, or hTERT in humans) is a subunit of the enzyme telomerase.
TERT inhibition has been linked directly or indirectly to all hallmarks of aging, but TERT expression is linked to the development of many cancers.
Unfortunately, TERT gene is epigenetically repressed with the onset of aging markers in all tissues.
Telomerase reverse transcriptase (abbreviated to TERT, or hTERT in humans) is a subunit of the enzyme telomerase.
TERT inhibition has been linked directly or indirectly to all hallmarks of aging, but TERT expression is linked to the development of many cancers.
Unfortunately, TERT gene is epigenetically repressed with the onset of aging markers in all tissues. Ribosomopathies encompass a wide range of syndromes. Common symptoms include a reduced number of blood cells, predisposition to cancer, skeletal abnormalities, and failure to thrive. Ribosomopathy has been associated with skeletal muscle atrophy, which brings us closer to ALS.
Ribosomopathies encompass a wide range of syndromes. Common symptoms include a reduced number of blood cells, predisposition to cancer, skeletal abnormalities, and failure to thrive. Ribosomopathy has been associated with skeletal muscle atrophy, which brings us closer to ALS. There is nothing new in the list of drugs the authors list, and they lump in the same basket many unrelated drugs.
There is nothing new in the list of drugs the authors list, and they lump in the same basket many unrelated drugs. Curtiss Neveu via Wikipedia
Curtiss Neveu via Wikipedia Les lymphocytes T matures sont considérés comme immunologiquement naïfs jusqu'à ce qu'ils rencontrent le peptide spécifique dans le contexte d'une molécule d'antigène leucocytaire humain (HLA) que leur récepteur reconnaît. Une fois la reconnaissance de l’antigène effectuée, les cellules reçoivent un signal prolifératif qui conduit à une expansion marquée des lymphocytes T spécifiques de l’antigène et à une réponse inflammatoire. Bien que beaucoup de ces cellules subissent l’apoptose après la réponse initiale, d’autres sont sauvées de la rétraction immunitaire et persistent sous forme de cellules T mémoire. Les lymphocytes T mémoire peuvent répondre rapidement à une nouvelle provocation spécifique d’un antigène et persister dans la circulation à long terme
Les lymphocytes T matures sont considérés comme immunologiquement naïfs jusqu'à ce qu'ils rencontrent le peptide spécifique dans le contexte d'une molécule d'antigène leucocytaire humain (HLA) que leur récepteur reconnaît. Une fois la reconnaissance de l’antigène effectuée, les cellules reçoivent un signal prolifératif qui conduit à une expansion marquée des lymphocytes T spécifiques de l’antigène et à une réponse inflammatoire. Bien que beaucoup de ces cellules subissent l’apoptose après la réponse initiale, d’autres sont sauvées de la rétraction immunitaire et persistent sous forme de cellules T mémoire. Les lymphocytes T mémoire peuvent répondre rapidement à une nouvelle provocation spécifique d’un antigène et persister dans la circulation à long terme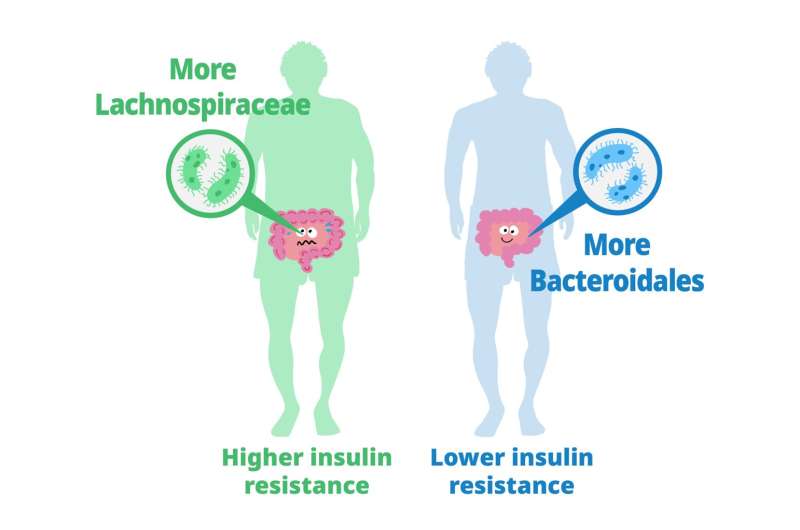 Previous studies have explored the role of gut microbiota in metabolizing nutrients in insulin resistance. This research aims to uncover the mechanisms underlying this relationship using a multi-omics approach. The study analyzes data from 306 individuals without diabetes, focusing on insulin resistance as defined by HOMA-insulin resistance scores.
Previous studies have explored the role of gut microbiota in metabolizing nutrients in insulin resistance. This research aims to uncover the mechanisms underlying this relationship using a multi-omics approach. The study analyzes data from 306 individuals without diabetes, focusing on insulin resistance as defined by HOMA-insulin resistance scores. Until now, studies on Klotho have primarily focused on its effects on animal models such as mice, rather than directly increasing its levels in primates.
Until now, studies on Klotho have primarily focused on its effects on animal models such as mice, rather than directly increasing its levels in primates. This condition affects individuals, mostly adults over the age of 50, who physically act out their dreams during sleep, resulting in injuries to themselves or their bed partners. It's also suspected to be involved in premisses of Parkinson's disease. The study, published in the Journal of Neuroscience, presents a novel model that characterizes how REM sleep behavior disorder develops due to neurodegeneration associated with the accumulation of tau protein.
This condition affects individuals, mostly adults over the age of 50, who physically act out their dreams during sleep, resulting in injuries to themselves or their bed partners. It's also suspected to be involved in premisses of Parkinson's disease. The study, published in the Journal of Neuroscience, presents a novel model that characterizes how REM sleep behavior disorder develops due to neurodegeneration associated with the accumulation of tau protein.