Des scientifiques Japonais, dont le célèbre spécialiste de la SLA Makoto Urushitani , ainsi que le premier auteur Ryutaro Nakamura, ont voulu étudier si l'hypermétabolisme, mais aussi le métabolisme des lipides, pouvait prédire le pronostic de patients atteints de sclérose latérale amyotrophique (SLA) précoce, ayant des profils nutritionnels différents lors de leur hospitalisation.

En effet de nombreux éléments montrent l'importance du traitement non pharmacologique qui peut améliorer la qualité de vie, diminuer la progression ou augmenter la durée de survie lors de la SLA.
L'introduction précoce de la ventilation à pression positive non invasive, en particulier avec les dispositifs d'assistance à la toux, prolongerait la durée de vie de la SLA.
De plus un régime riche en calories ralentit efficacement la progression de la SLA, en particulier chez les patients à progression rapide.
Étonnamment, dans la SLA à progression rapide, un régime hypercalorique à dominance grasse inhibe l'élévation du neurofilament-L (NFL) sérique, un biomarqueur convaincant des dommages neuronaux.
Un indice de masse corporelle (IMC) inférieur à 18,5 lors de la première visite chez les neurologues est reconnu comme un facteur de mauvais pronostic.
Malgré ces éléments cliniques, et bien que de nombreuses études aient montrées que l’âge et l’IMC sont les meilleurs marqueurs biologiques pour prédire l’évolution de la SLA, ces études se heurtent à une sorte de barrière narrative consensuelle qui dit que la SLA est une maladie des neurones moteurs supérieurs et que la perte de masse musculaire est exclusivement due à l’absence d’activation par les neurones moteurs inférieurs.
De façon similaire alors que les scientifiques disent qu’une protéopathie de type TDP-43 est présente dans l’ensemble des cellules du corps des malades, particulièrement dans les muscles squelettiques (ceux impliqués dans la SLA) dans 97 % des cas, il semble bien improbable que la SLA ne ciblerait que certaines cellules et uniquement dans le cerveau et la moelle épinière.
Enfin, plus de la moitié des patients de la SLA sont atteints d’hypermétabolisme, c’est-à-dire que pour effectuer une certaine tâche, leur corps a besoin de davantage d’énergie que le corps d’une personne en bonne santé.
Les patients atteints de SLA ont une tolérance au glucose qui est altérée dès un stade précoce, et les acides gras libres sont plus élevés chez ces patients que chez ceux ayant une tolérance normale au glucose. Il a été rapporté que la voie de production d'énergie passe du métabolisme glycolytique au métabolisme lipidique au cours de la maladie.
Cela indique que les lipides pourraient être le principal carburant chez les patients souffrant de malnutrition ainsi que chez les souris SLA, bien qu'aucune étude n'ait examiné les changements du métabolisme en réponse à l'état nutritionnel des humains.
Des taux élevés de cholestérol total et de cholestérol à lipoprotéines de basse densité (LDL) et un rapport LDL/cholestérol à lipoprotéines de haute densité (HDL) plus élevé ont été associés à une survie plus longue, ce qui pourrait indiquer un métabolisme utilisant principalement les lipides plutôt que le glucose.
Par conséquent, Ryutaro Nakamura et ses collègues ont examiné rétrospectivement les informations disponibles sur quarante-huit patients (25 hommes ; 23 femmes) de plus de 49 ans, atteints de SLA et admis à l'hôpital de l'Université des sciences médicales de Shiga de mars 2018 à février 2021.
Le but des scientifiques était de tester l'hypothèse selon laquelle l'impact de l'hypermétabolisme sur la survie pourrait différer selon l'état nutritionnel.
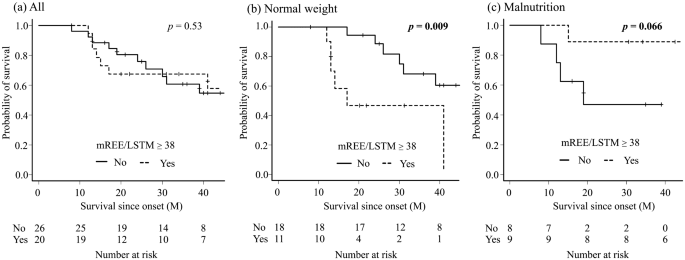
Fig. 1: Comparaison du taux de survie entre le groupe hypermétabolique et le groupe métabolique normal chez tous les patients (a), le groupe de poids normal (b), le groupe de malnutrition (c). Les analyses de Kaplan-Meier et le test du log-rank ont montré qu'il n'y avait pas de différence chez tous les patients ; cependant, le groupe hypermétabolique avait un temps de survie significativement plus court dans le groupe de poids normal et un temps de survie plus long dans le groupe malnutrition, bien que la différence ne soit pas significative.
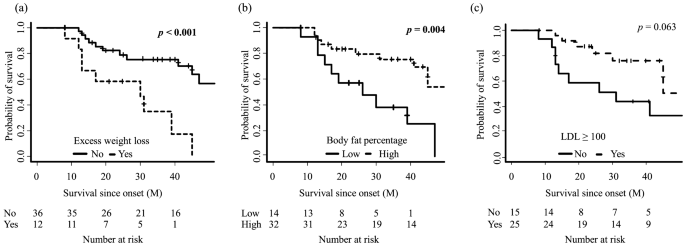
Fig. 2: Comparaison du taux de survie entre les patients atteints de sclérose latérale amyotrophique stratifiée par perte de poids excessive (a), pourcentage de graisse corporelle (b) et cholestérol à lipoprotéines de basse densité (LDL) (c). les analyses ont montré que les patients avec une perte de poids excessive et un faible pourcentage de graisse corporelle avaient un temps de survie significativement plus court.
Dans cette étude, les scientifiques Japonais ont exploré l'effet de l'hypermétabolisme sur la survie par rapport à l'état nutritionnel des patients atteints de SLA, et ont constaté que les patients hypermétaboliques avec un poids normal avaient une survie plus courte, alors que la survie était plus longue chez les patients hypermétaboliques souffrant de malnutrition. Ils constatent que la survie est corrélée avec le taux de LDL.
Le niveau faible de muscle squelettique et de masse des tissus mous, qui est un facteur de mauvais pronostic, pourraient être la raison pour laquelle le pronostic du groupe hypermétabolique de poids normal est plus mauvais que celui des autres groupes.
Étonnamment, l'hypermétabolisme était un meilleur facteur pronostique dans le groupe malnutrition (Fig. 1). Les biomarqueurs lipidiques étaient caractéristiques dans ce groupe, ce qui pourrait contribuer à une survie plus longue.
Le groupe de malnutrition hypermétabolique avait la valeur médiane de LDL la plus élevée parmi tous les groupes, et un pourcentage de graisse corporelle plus élevé, qui étaient de bons facteurs pronostiques dans les études précédentes.
En effet, les patients avec un pourcentage de graisse corporelle élevé avaient une survie plus longue dans leur étude (Fig. 2), et les patients avec un LDL élevé ont montré une tendance similaire.
Il convient de noter que le quotient respiratoire était inférieur à 0,85 chez huit des neuf patients de ce groupe et que la glycémie à jeun était significativement plus élevée dans ce groupe, ce qui pourrait indiquer un changement du métabolisme lipidique par rapport à l'hypothèse de glucose.
Le quotient respiratoire était associé à l'ALSFRS-R et au temps écoulé depuis le début, et il est intéressant d'émettre l'hypothèse qu'un « commutateur de carburant » puisse sous-tendre les profils métaboliques dans la progression de la SLA.
De plus, Steyn et al. ont rapporté qu'une oxydation élevée des acides gras augmente la dépense énergétique du corps entier et ralentit la progression de la maladie, ce qui pourrait étayer les résultats décrits ici et indiquer la contribution du métabolisme hyperlipidique à une survie plus longue. ces résultats mettent en garde contre la mesure de l'hypermétabolisme comme seul indicateur de pronostic.
Sur la base de l'implication possible de l'hypermétabolisme des muscles squelettiques dans le pronostic de la SLA, les scientifiques Japonais proposent l'équation suivante «BMM = (IMC − 19,8) × (mREE/LSTM − 38) », qui a prédit avec succès le facteur pronostique, quels que soient les états nutritionnels.
BMM = BMI-métabolisme musculaire
mREE = dépense énergétique au repos mesurée
LSTM = masse maigre des tissus mous
En conclusion, ces travaux démontrent qu'une perte de poids de plus de 10 % était un facteur de mauvais pronostic. Le taux de perte de poids était plus rapide dans le groupe à indice BMM élevé ; ainsi, l'indice BMM pourrait être un indicateur précieux pour envisager une intervention nutritionnelle agressive, y compris le PEG (gastrostomie endoscopique percutanée), à un stade précoce de la pratique clinique.
La supplémentation calorique riche en graisses ou en glucides est encore un sujet de débat.
Certains rapports suggèrent que la supplémentation en calories riches en graisses est un meilleur traitement nutritionnel chez les patients atteints de SLA, tandis que d'autres recommandent une supplémentation en calories riches en glucides.
Pour les patients atteints de maladie pulmonaire obstructive chronique qui ont des émissions de CO2 réduites, un régime riche en graisses est recommandé car le métabolisme des lipides produit moins de dioxyde de carbone. Sur la base de leur résultat selon lequel la tendance à la modification des lipides pourrait contribuer à un meilleur pronostic, un apport riche en graisses pourrait être une option attrayante dans un régime riche en calories pour la SLA. Bien entendu, le choix de la nutrition doit être fait avec prudence, en fonction des profils métaboliques des patients.
La définition que les auteurs ont adoptée pour l'hypermétabolisme est un mREE/LSTM élevé, plutôt que l'indice mondial populaire, mREE/pREE, car le PRE estimé par l'équation de Harris-Benedict ne serait pas approprié pour l'évaluation métabolique des Japonais.
 By Jynto via Wikipedia
By Jynto via Wikipedia
 By National Human Genome Research Institute - via Wikipedia
By National Human Genome Research Institute - via Wikipedia
 Source Wikipedia: FrozenMan - Own work
Source Wikipedia: FrozenMan - Own work
