Normalement, je n'envisagerais pas d'inclure des articles qui n'impliquent pas de traitements humains ou qui utilisent une méthodologie médiocre telle que l'enrichissement GO, mais cet article est intéressant car les mouvements y sont restaurés dans un modèle animal de SLA.
Si nous suivons le consensus scientifique actuel, cela serait impossible car les neurones moteurs meurent et ils ne sont pas remplacables, mais certains patients SLA prétendent avoir, au moins en partie, récupéré une certaine capacité. Moins du quart des publications scientifiques sur la SLA discutent de l'apparition des défauts dans les muscles squelettiques avant même l'apparition d'anomalies des motoneurones. Il s'agit de l'hypothèse dite du «dépérissement» qui énonce que les problèmes apparaissent au niveau musculaire et se transmettent aux neurones, alors que l'hypothèse majoritaire est calquée depuis 150 ans sur le modèle de l'AVC et où les neurones moteurs supérieures meurent (leur corps est dans la zone motrice du cerveau), puis faute d'activation les moteurs neurones inférieurs meurent et à leur tour les muscles meurent. Cette hypothèse pourtant consensuelle est cependant difficile à concilier avec les formes de SLA où seuls les neurones moteurs inférieurs seraient touchés. En particulier un neurone moteur dont les synapses seraient non fonctionnels ou inhibés serait indiscernable d'un neurone moteur mort, dans la mesure où les techniques d'observation utilisées sont toutes indirectes.
Je discute également içi de cet article parce que des essais cliniques sur la SLA portant sur des médicaments proches de ceux suggérés dans cet article ont montré un léger ralentissement de la progression de la maladie. Hélas de nombreux essais cliniques ont montrés des effets similaires mais insuffisants pour obtenir un agrément de mise sur le marché.
Les inclusions de TDP-43 dans les cellules cérébrales sont une caractéristique commune de nombreuses maladies neurodégénératives, dont la SLA et la FTD. Le texte souligne avec justesse que si les mutations de TDP-43 expliquent une petite partie des cas, divers facteurs peuvent conduire à l'agrégation de granules de TDP-43 dans le cytosol, au lieu du noyau des cellules, qui est censée provoquer une neurodégénérescence.
Les auteurs mentionnent plusieurs processus pathogènes liés à cela, tels que l'excitotoxicité induite par le glutamate, la dérégulation du métabolisme de l'ARN, l'altération de la dégradation des protéines et le dysfonctionnement mitochondrial.
Le texte discute de la nécessité d'élargir le champ de la recherche qui est actuellement concentrée sur les facteurs intrinsèques aux cellules et de la nécessité de considérer l'impact sur les réseaux synaptiques. C'est un point de vue que je défends aussi. Le système nerveux est un réseau complexe et interconnecté et que la fonction d’un neurone dépend de ses interactions avec d’autres neurones et aussi avec les systèmes musculaire et sensoriel. Si le réseau est perturbé, les interventions axées uniquement sur des neurones individuels pourraient ne pas suffire à rétablir leur bon fonctionnement. Au lieu de cela, rétablir l’équilibre et une connectivité adéquate au sein du réseau pourrait conduire à de meilleurs résultats en termes de fonction motrice.
Pour étudier le rôle des neurones malades dans un ensemble de réseaux nerveux et musculaires, les auteurs se sont tourné vers le nématode Caenorhabditis elegans. Le nématode Caenorhabditis elegans, possède un système nerveux de seulement 302 cellules, un connectome entièrement établi et des systèmes de neurotransmetteurs comparables à ceux des humains, fournit un modèle simplifié pour une approche systémique. Ils décrivent comment la surexpression du TDP-43 humain dans ce modèle conduit à une toxicité cellulaire et à une paralysie, imitant les caractéristiques de la SLA.
La section des résultats se penche sur le profilage comportemental et identifie la transmission du GABA et de l'acétylcholine comme les processus clés responsables des phénotypes induits par le TDP-43. Cette approche est utilisée pour évaluer comment les gènes, les facteurs environnementaux et les mécanismes neuronaux influencent le comportement d'un organisme. Le phénotypage comportemental implique généralement l'observation, la mesure et la quantification d'un large éventail de comportements, tels que l'activité locomotrice, les interactions sociales, l'apprentissage, la mémoire et les réponses aux facteurs de stress. Cette empreinte digitale a été comparée à celle de 294 mutants de C. elegans, dans lesquels ont été mutés un large spectre de gènes importants pour le fonctionnement du système nerveux et des muscles. En utilisant une approche de regroupement informatique, les auteurs ont constaté que la libération d’acétylcholine et de GABA était le principal défaut des nématodes modèle de la SLA. Il faut cependant bien garder à l'esprit que tous les modèles sont imparfaits, et que les animaux modèles de la SLA sont des modèles très imparfaits.
Le texte explore les dommages différentiels causés aux neurones GABAergiques et cholinergiques, le rôle de l'hypoexcitabilité intrinsèque et la modification du connectome. Les scientifiques proposent un modèle suggérant que la toxicité du TDP-43 provoque des modifications du rapport E/I du muscle, ce qui affecte la fonction motrice. Les neurones coordonnent leurs entrées excitatrices et inhibitrices pour établir et maintenir un rapport excitation/inhibition (E/I) constant, considéré comme essentiel au fonctionnement et à la stabilité de la fonction.
Les preuves expérimentales soutiennent l'idée selon laquelle une excitation et une inhibition équilibrées au sein des circuits neuronaux facilitent leur fonctionnement et que le manque de maintien de l'équilibre E/I est à l'origine du dysfonctionnement des circuits observé dans de nombreuses maladies neurologiques.
Les muscles de la paroi corporelle de C. elegans reçoivent des entrées de motoneurones GABAergiques excitateurs et cholinergiques au niveau des jonctions neuromusculaires (NMJ), ce qui en fait un excellent modèle pour étudier les mécanismes génétiques et moléculaires nécessaires au maintien de l'équilibre E-I au niveau du NMJ.
Les auteurs du texte ont pu restaurer les mouvements dans le modèle de C. elegans pour la toxicité induite par le TDP-43 grâce à plusieurs interventions visant à rééquilibrer le rapport excitation/inhibition (E/I) dans le circuit moteur. Ils ont exploré différents scénarios et voici comment ils ont réussi à restaurer le mouvement :
Inhibition de la signalisation cholinergique : ils ont partiellement inhibé la signalisation cholinergique de manière post-synaptique. Cette approche a déplacé l’équilibre E/I vers l’inhibition. Cela a été fait en traitant les vers avec le dTBC, un antagoniste de l'AChR, ce qui a entraîné une augmentation significative de la fréquence des raclées.
Amélioration de la signalisation cholinergique : Ils ont émis l’hypothèse que l’amélioration de la signalisation cholinergique, ainsi que l’inhibition post-synaptique, pourraient encore augmenter la capacité de mouvement des vers. Ils ont utilisé l'inhibition pharmacologique des gènes post-synaptiques ou l'inhibition transcriptionnelle et ont traité les vers avec l'arécoline, un activateur de Gq, qui a considérablement augmenté la capacité de mouvement.
Stimulation directe de la signalisation GABAergique : Ils ont directement stimulé les GABAR (récepteurs GABA) avec du muscimol.
Approche combinée : Ils ont combiné l'amélioration de la signalisation cholinergique et GABAergique simultanément mais à des ampleurs différentes, ce qui a permis de maximiser la capacité de mouvement des vers hTDP-43.
Ces interventions ont été conçues pour rééquilibrer le rapport E/I perturbé dans le circuit moteur provoqué par la toxicité du TDP-43. Les résultats suggèrent que le rétablissement de l’équilibre des circuits neuronaux, plutôt que la simple amélioration des systèmes de neurotransmetteurs individuels, sont cruciaux pour restaurer la fonction motrice dans les maladies neurodégénératives.
il existe plusieurs médicaments qui modulent les récepteurs cholinergiques et GABAergiques, et nombre d'entre eux sont utilisés pour diverses conditions médicales. Voici quelques exemples:
Médicaments cholinergiques : Inhibiteurs de la cholinestérase, agonistes muscariniques, agonistes nicotiniques.
Médicaments GABAergiques : Benzodiazépines, barbituriques, gabapentine et prégabaline, agonistes des récepteurs GABA.
Bien qu'il n'existe aucun essai clinique sur une intervention combinant des médicaments cholinergiques et GABAergiques, des interventions cholinergiques ont été testées dans des essais cliniques : une supplémentation orale en um-PEA (Palmitoyléthanolamide), chez un patient atteint de SLA, a conduit à une amélioration du tableau clinique, du tonus musculaire. , respiratoires et motrices, grâce au contrôle PEA de la neuroinflammation [51]. Plus récemment, un vaste essai clinique sur des patients atteints de SLA a démontré que l'administration pendant 6 mois d'um-PEA, en plus du traitement standard (riluzole), ralentissait le déclin de la fonctionnalité pulmonaire et l'aggravation des symptômes de la SLA.
Il y a eu au cours des années précédentes un essai clinique sur la mexilétine dans la sclérose latérale amyotrophique sporadique qui a montré certains effets.
Il serait intéressant de faire ce type de recherche sur des primates modèles de la maladie. Mais il sera difficile de trouver un financement pour ce type de recherche qui contredit le dogme fondamentale de la recherche dans la SLA qui veut que sur le modèle de l'AVC, ce soit les neurones moteurs supérieurs qui meurent d'abord.
 Yichang Jia, Ph.D., Co-founder of SineuGene
Yichang Jia, Ph.D., Co-founder of SineuGene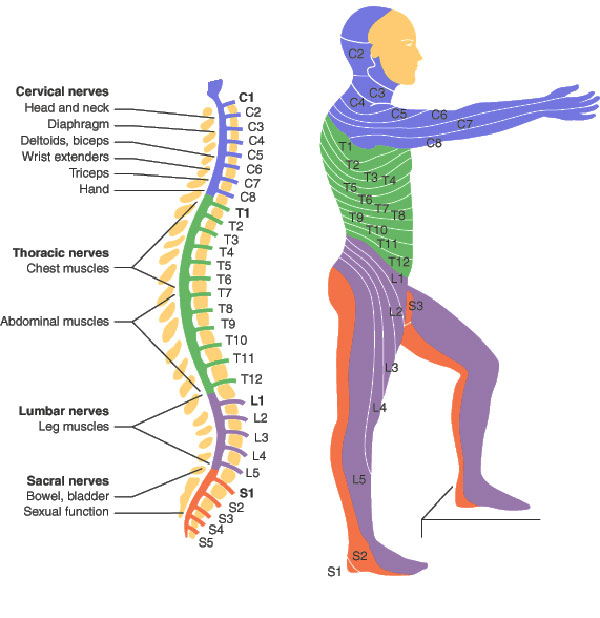 Obviously, a spinal cord injury will sever the link between the brain motor area, some upper motor neurons, and corresponding lower motor neurons and muscles. Yet it does not stop there, which is particularly interesting when we have ALS in mind. In spinal-onset ALS the disease starts in a very localized muscular for example a muscle in the thumb, and it spreads, often until respiratory muscles fail.
Obviously, a spinal cord injury will sever the link between the brain motor area, some upper motor neurons, and corresponding lower motor neurons and muscles. Yet it does not stop there, which is particularly interesting when we have ALS in mind. In spinal-onset ALS the disease starts in a very localized muscular for example a muscle in the thumb, and it spreads, often until respiratory muscles fail.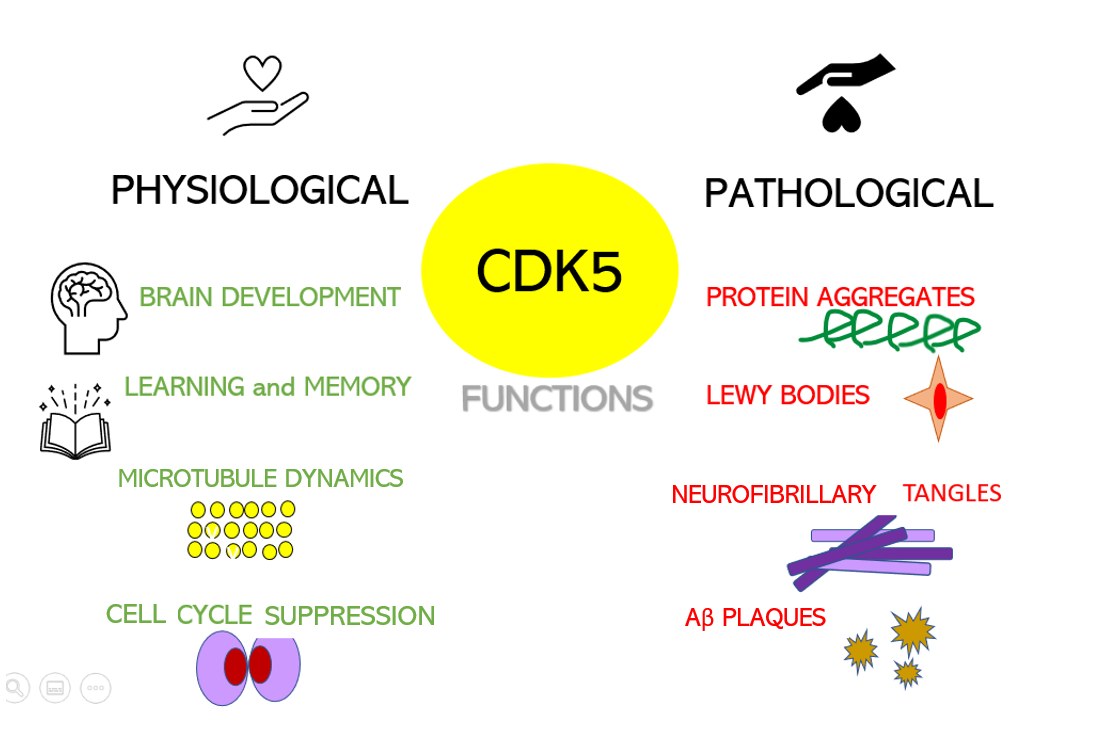 Dysregulation of CDKs, particularly cyclin-dependent kinase 5 (Cdk5), is seen in many neurological disorders, including Alzheimer's disease (AD) and Parkinson's disease (PD). Cdk5 is a unique member of the CDK family because it does not play a critical role in cell cycle progression and is not activated by a cyclin. Instead, Cdk5 is normally activated by the regulatory protein p25. Cdk5/p35/p25 activity is normally an important regulator of the proper development of the mammalian central nervous system.
Dysregulation of CDKs, particularly cyclin-dependent kinase 5 (Cdk5), is seen in many neurological disorders, including Alzheimer's disease (AD) and Parkinson's disease (PD). Cdk5 is a unique member of the CDK family because it does not play a critical role in cell cycle progression and is not activated by a cyclin. Instead, Cdk5 is normally activated by the regulatory protein p25. Cdk5/p35/p25 activity is normally an important regulator of the proper development of the mammalian central nervous system. Their results show that the model can identify, from 3 seconds of magnetoencephalographic signals, the corresponding speech segment with up to 41% accuracy, which is, however, lower than previous results. Furthermore, we know that
Their results show that the model can identify, from 3 seconds of magnetoencephalographic signals, the corresponding speech segment with up to 41% accuracy, which is, however, lower than previous results. Furthermore, we know that  If I extrapolate from Figure 6, patients with two treatments would live 13 years longer than patients without treatment. This improvement is considerable, if we ignore the statistical hallucinations of pharmaceutical companies, the current improvements simply relate to a few months of survival. We will see below that we can doubt this result.
If I extrapolate from Figure 6, patients with two treatments would live 13 years longer than patients without treatment. This improvement is considerable, if we ignore the statistical hallucinations of pharmaceutical companies, the current improvements simply relate to a few months of survival. We will see below that we can doubt this result. Any way at least if it works in human patients with FUS/C9orf72 that would mean one in five ALS patients would benefit from it. This would be much larger than Qualsody benefits.
Any way at least if it works in human patients with FUS/C9orf72 that would mean one in five ALS patients would benefit from it. This would be much larger than Qualsody benefits. There was a reduced risk of death observed in patients exposed to a higher dosage (defined as ≥ 1000 mg/day) of TUDCA compared to both the control group and those with lower TUDCA dosages.
There was a reduced risk of death observed in patients exposed to a higher dosage (defined as ≥ 1000 mg/day) of TUDCA compared to both the control group and those with lower TUDCA dosages.