In this blog, I avoid studies that are not done on humans, firstly because the further away from humans and primates, the less sincere the scientific studies are. Then it is well known that the pre-clinical studies published are all extremely positive in order to attract investors (if possible private) who will finance clinical studies.
2,4-dinitrophenol (DNP)
The study examined here explores whether small doses of 2,4-dinitrophenol, a chemical used in the manufacture of pesticides and slimming drugs because it decreases the metabolic action of mitochondria, could help protect motor neurons, preserve muscle function and slow the progression of amyotrophic lateral sclerosis (ALS) in a mouse model of the disease. The scientists' guiding idea therefore seems to be to slow down the disease, by slowing down the metabolism. This seems absolutely counter-intuitive but the scientists assure that they have had good results. The effective dose of 2,4-dinitrophenol was very low (0.5–1 mg/kg, human equivalent dose 2.5–5 mg/day), making it safer for potential use in humans.
 Dinitrophenol acts as a proton transporter in the mitochondrial membrane, inhibiting oxidative phosphorylation of ATP and making energy production less efficient. This is because some of the energy that is normally produced from cellular respiration is wasted as heat. This inefficiency is proportional to the dose of dinitrophenol that is absorbed. Thus, as the dose increases, energy production becomes less efficient: metabolism is then activated - more fat is burned - to compensate for the inefficiency and meet energy demands.
Dinitrophenol acts as a proton transporter in the mitochondrial membrane, inhibiting oxidative phosphorylation of ATP and making energy production less efficient. This is because some of the energy that is normally produced from cellular respiration is wasted as heat. This inefficiency is proportional to the dose of dinitrophenol that is absorbed. Thus, as the dose increases, energy production becomes less efficient: metabolism is then activated - more fat is burned - to compensate for the inefficiency and meet energy demands.
The researchers used hSOD1G93A mice, a common model of ALS, to test the effects of 2,4-dinitrophenol on motor skills, muscle strength, and disease progression. However, it is important to remember that most ALS patients do not have this mutation and that its notoriety is simply because it was the first mutation to be associated with ALS and that for more than 10 years (from 1993 to 2006) no other deleterious mutations were discovered in ALS.
Results
Mice treated with microdoses of 2,4-dinitrophenol (0.5–1 mg/kg) showed better motor coordination and better results on tests measuring muscle strength, compared to untreated mice. Early treatment initiation (before symptoms appeared) delayed the onset of motor decline, while late treatment initiation (after symptoms appeared) improved motor skills (which were already impaired by the disease) and slowed disease progression. Treated mice retained their ability to perform tasks such as running at 20 cm/s for longer than untreated diseased mice. In some cases, the mice regained lost motor skills, which is unusual in ALS research.
Although ALS damages the connections between nerves and muscles (neuromuscular junctions), treatment with 2,4-dinitrophenol preserved these connections. Treated mice retained a higher number of motor units (groups of muscle fibers controlled by a single neuron), indicating that motor neuron loss was reduced.
Reduced Cellular Stress
2,4-dinitrophenol reduced oxidative stress, a harmful process linked to the progression of ALS. This was demonstrated by lower levels of damaged proteins in treated muscles. While slowing down an ALS patient’s metabolism seems pretty criminal, reducing cellular stress is a very good idea.
The drug also reduced inflammation and activated pathways (like Akt/mTOR) involved in muscle growth and repair.
How would 2,4-dinitrophenol work?
2,4-dinitrophenol acts on mitochondria, the energy producers of cells, by slightly lowering their membrane potential (∆Ψm). This mild “uncoupling” reduces the production of harmful reactive oxygen species (ROS), which can damage cells. 2,4-dinitrophenol may also help clear damaged mitochondria and promote the formation of healthy mitochondria, further protecting neurons and muscles.
Future Directions
While the mouse results are promising, translating these findings to humans requires more research. The study also highlighted some limitations:
It focused on one type of ALS model (hSOD1G93A) that affects less than 2% of patients.
It did not address potential differences in drug responses between male and female mice, which could be significant.
Administering a metabolic reducer to ALS patients still seems like a dangerous notion.
 Angel Bu is the first author, while Ritu Raman is the senior author; the other authors are from MIT’s Department of Mechanical Engineering and MIT’s Koch Institute for Integrative Cancer Research. The authors matured a set of motor neurons on a gel that was a kind of carpet into which they embedded tiny magnets. They then used an external magnet to shake the carpet—and the neurons—back and forth. In this way, they made the neurons work, for 30 minutes a day.
Angel Bu is the first author, while Ritu Raman is the senior author; the other authors are from MIT’s Department of Mechanical Engineering and MIT’s Koch Institute for Integrative Cancer Research. The authors matured a set of motor neurons on a gel that was a kind of carpet into which they embedded tiny magnets. They then used an external magnet to shake the carpet—and the neurons—back and forth. In this way, they made the neurons work, for 30 minutes a day. Early immunotherapies targeting amyloid-β in Alzheimer's disease initially showed reductions in amyloid plaques but failed to prevent cognitive decline. The FDA recently approved lecanemab as the first disease-modifying drug for Alzheimer's disease. However, its benefits appear to be limited to the early stages of the disease, as it does not stop neurodegeneration or improve cognition. This highlights the difficulty of modifying neurodegenerative diseases, which often progress despite treatment.
Early immunotherapies targeting amyloid-β in Alzheimer's disease initially showed reductions in amyloid plaques but failed to prevent cognitive decline. The FDA recently approved lecanemab as the first disease-modifying drug for Alzheimer's disease. However, its benefits appear to be limited to the early stages of the disease, as it does not stop neurodegeneration or improve cognition. This highlights the difficulty of modifying neurodegenerative diseases, which often progress despite treatment. Curiously, scientists are rather looking to directly convert astrocytes into neurons, despite the enormous morphological difference between these two types of cells.
Curiously, scientists are rather looking to directly convert astrocytes into neurons, despite the enormous morphological difference between these two types of cells.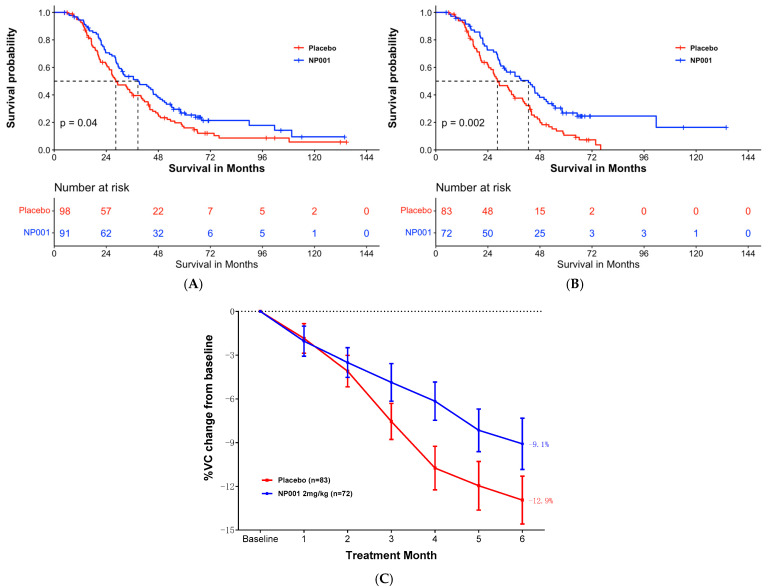 For example, the authors claim that survival of ALS patients with inflammation is 16 months longer than in the placebo arm. At 72 months there were only 3 people in the inflammation subset and two people in the placebo arm. Nobody can say anything about these numbers. If we use the same criteria, it shows that NP001 worsened the condition in all pALS with respect to the placebo branch, as starting from 72 months there were fewer survivors in the NP001 arm than in the placebo.
For example, the authors claim that survival of ALS patients with inflammation is 16 months longer than in the placebo arm. At 72 months there were only 3 people in the inflammation subset and two people in the placebo arm. Nobody can say anything about these numbers. If we use the same criteria, it shows that NP001 worsened the condition in all pALS with respect to the placebo branch, as starting from 72 months there were fewer survivors in the NP001 arm than in the placebo. The authors observed that, despite a decrease in limb motor functions, the feeding function of these mice was preserved until the late stages of the disease. Remarkably, the masseter muscle showed no reduction in muscle volume, wet weight, or muscle fiber cross-sectional area. Furthermore, no changes were observed in muscle fiber types, indicating a possible resistance of the masseter muscle to ALS-induced impairment. A potential reason for the lack of atrophy in the masseter muscle could be its higher number of muscle satellite cells compared to that of the gastrocnemius muscle. This abundance may promote the maintenance of muscle fiber nuclei, thereby contributing to muscle tissue regeneration.
The authors observed that, despite a decrease in limb motor functions, the feeding function of these mice was preserved until the late stages of the disease. Remarkably, the masseter muscle showed no reduction in muscle volume, wet weight, or muscle fiber cross-sectional area. Furthermore, no changes were observed in muscle fiber types, indicating a possible resistance of the masseter muscle to ALS-induced impairment. A potential reason for the lack of atrophy in the masseter muscle could be its higher number of muscle satellite cells compared to that of the gastrocnemius muscle. This abundance may promote the maintenance of muscle fiber nuclei, thereby contributing to muscle tissue regeneration.