Alzheimer's disease (AD) is a progressive and irreversible neurodegenerative brain disorder. Despite its prevalence and severity, early diagnosis of AD remains a considerable challenge.
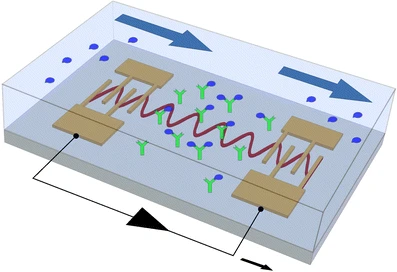
Scientists from Duke University, report an integrated acoustofluidics-based diagnostic system (ADx), which combines triple functions of acoustics, microfluidics, and orthogonal biosensors for clinically accurate, sensitive, and rapid detection of AD biomarkers from human plasma.
They also designed and built a surface acoustic wave-based acoustofluidic separation device to isolate and purify Alzheimer's disease biomarkers to increase the signal-to-noise ratio.
Semiconductor metal oxide nanostructures have various compositions and morphologies such as single crystals, one dimensional, and thin or thick film form. Recently, 1D nanoarchitectors have much attraction for sensing due to their aspect ratio beside their thermal and chemical stabilities.
Nanostructures grown by these techniques could adopt the shape of nanowires, nanobelts, nanotubes, nanoneedles, nanorods, etc...
The modifications in surface morphology result in sensing of various kinds of oxidizing and reducing gases like CO, NH3, NO2, H2, O2, H2S, LPG, xylene, propane, toluene, triethylamine, methanol, and acetone.
Multimodal biosensors within the integrated acoustofluidics-based Alzheimer disease diagnostic system were fabricated by in-situ patterning of the ZnO nanorod array and deposition of Ag nanoparticles onto the ZnO nanorods for surface-enhanced Raman scattering (SERS) and electrochemical immunosensors.
This diagnostic system enabled the label-free detections of SERS and electrochemical immunoassay of clinical plasma samples from AD patients and healthy controls with high sensitivity and specificity.
The researchers believe that this efficient integration provides promising solutions for the early diagnosis of AD.
 Source: KieranMaher at English Wikibooks
Source: KieranMaher at English Wikibooks
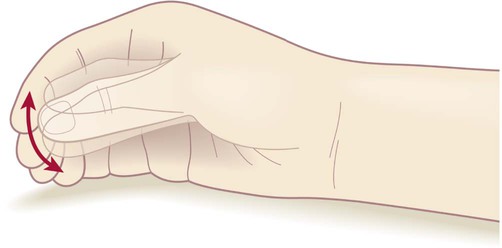 ‘Pill-rolling’ rest tremor as found in Parkinson’s disease.
‘Pill-rolling’ rest tremor as found in Parkinson’s disease.