Ce blog traite principalement des trois principales maladies neurodégénératives, Alzheimer, Parkinson et SLA. Malheureusement, dans la vie réelle, les maladies neurodégénératives n'ont pas de symptômes bien différenciés.
Lorsque Parkinson et SLA sont souvent associés à la démence, il est moins courant d'associer Parkinson et Autisme.
 Un récent article de prépublication sur MedRxiv, rédigé par Kadalraja Raghavan, Samuel JK Abraham et ses collègues d'Inde et du Japon, indique qu'un régime dérivé d'un champignon noir a pu atténuer les symptômes de l'autisme chez les enfants présentant des symptômes graves, et encore plus surprenant, il a abaissé leur niveau d'α-synucléine dans le sang.
Un récent article de prépublication sur MedRxiv, rédigé par Kadalraja Raghavan, Samuel JK Abraham et ses collègues d'Inde et du Japon, indique qu'un régime dérivé d'un champignon noir a pu atténuer les symptômes de l'autisme chez les enfants présentant des symptômes graves, et encore plus surprenant, il a abaissé leur niveau d'α-synucléine dans le sang.
Aucun mécanisme, intervention ou thérapie n'a prouvé sa capacité à réguler les niveaux d'α-synucléine. Dans cette étude, les taux plasmatiques d'α-synucléine ont montré une augmentation significative après la supplémentation en Nichi Glucan.
La maladie de Parkinson est une maladie neurodégénérative progressive caractérisée par des caractéristiques motrices cardinales, notamment des tremblements au repos, une rigidité, une instabilité posturale et une bradykinésie.
La maladie de Parkinson peut également présenter un large éventail de symptômes non moteurs, tels qu'un dysfonctionnement autonome, des troubles cognitifs, des troubles du sommeil et des symptômes neuropsychiatriques, notamment la dépression, l'anxiété et les comportements répétitifs ou obsessionnels compulsifs.
La maladie de Parkinson est caractérisée par des inclusions intracellulaires comprenant des dizaines de protéines mais principalement de l'α-synucléine. Les neurones dopaminergiques avec leurs longs axones disproportionnés qui se projettent de la substantia nigra (SN) dans le striatum sont très vulnérables à ces agrégats de protéines.
Les troubles du spectre autistique sont eux, caractérisés par des troubles de l'interaction sociale et des comportements répétitifs et stéréotypés. Les caractéristiques supplémentaires qui peuvent accompagner les troubles du spectre autistique sont les anomalies motrices, les problèmes gastro-intestinaux, l'épilepsie, la déficience intellectuelle ou les troubles du sommeil.
Bien que l'α-synucléine soit une protéine intracellulaire majoritairement localisée dans le cerveau, plusieurs études ont rapporté sa présence dans le plasma ainsi que dans le liquide céphalo-rachidien (LCR). Les niveaux d'α-synucléine sont connus pour être significativement plus bas chez les patients atteints de troubles du spectre autistique que chez les témoins sains.
La mesure de l'α-synucléine dans le LCR donnerait des résultats plus précis, mais cette procédure est invasive et n'est pas idéale pour une surveillance de routine.
Dans ce nouvel essai clinique pilote prospectif ouvert comprenant deux groupes, six enfants atteints de TSA ont reçu un traitement conventionnel comprenant des thérapies comportementales correctives et de la L-carnosine à raison de 500 mg par jour, et 12 enfants ont reçu une supplémentation de 0,5 g de Nichi Glucan deux fois par jour en plus du traitement conventionnel. Il n'y avait qu'un seul sujet féminin dans les deux groupes. L'étude a duré 90 jours. Un seul enfant a présenté des effets indésirables légers possibles liés à une augmentation des selles pendant une semaine.
Les sujets inclus dans l'étude avaient reçu un diagnostic de TSA par un pédiatre du développement, diagnostic vérifié par un psychologue à l'aide d'un entretien clinique pour un modèle comportemental qui incorporait l'échelle d'évaluation de l'autisme infantile (CARS).
Les quatres enfants du groupe témoin appartenaient à la catégorie de l'autisme sévère et leur score moyen au départ était de 42,75 ± 5,76. Parmi les neuf enfants ayant consommé du Nicho glucan, deux d'entre eux étaient atteint d'autisme léger à modéré (moyenne = 33,5 ± 2,5), tandis que les sept autres étaient dans la catégorie d'autisme sévère (moyenne = 43,71 ± 4,80). Après l'intervention, le score CARS moyen chez les quatre enfants du groupe témoin était pratiquement inchangé à 42,5 ± 5,4.
Dans le groupe ayant consommé du Nichi Glucan, la moyenne du score CARS chez les deux enfants atteints d'autisme léger à modéré était inchangée à 32,5 ± 0,5, tandis que chez les sept autres enfants, le score CARS s'améliorait légèrement (moyenne = 40,1 ± 5,96).
Dans ce dernier groupe, il y avait une réduction subjective visible de l'irritabilité et de la colère, une amélioration du sommeil, des caractéristiques de la parole et des réponses améliorées au soignant.
L'α-synucléine a récemment été considérée comme l'un des biomarqueurs importants pour le diagnostic de l'autisme et des TSA, où les niveaux sont faibles par rapport aux témoins du même âge (Kadak et al., 2015 ; Sriwimol et al., 2018 ; Siddique et al. 2020). Les taux plasmatiques d'α-synucléine étaient significativement plus élevés dans le groupe ayant consommé du Nichi Glucan que dans le groupe témoin.
Après l'intervention, les taux plasmatiques d'α-synucléine avaient en effet augmenté en moyenne de 26,72 ng/dl. Les taux d'α-synucléine humaine dans le plasma ont été mesurés dans le sang périphérique au départ et à la fin de l'étude qui durait 90 jours.
Les β-glucanes sont des polysaccharides naturellement présents dans les parois cellulaires des céréales, des bactéries et des champignons. Les β-glucanes trouvés dans les parois cellulaires de la levure contiennent un squelette de carbone 1,3 avec des branches de carbone 1,6 allongées. Nichi Glucan, est un complément alimentaire contenant du β-1,3-1,6-Glucan produit par la levure noire (souche AFO-202).
À des niveaux d'apport alimentaire d'au moins 3 g par jour, le β-glucane de fibres d'avoine diminue les taux sanguins de cholestérol LDL et peut ainsi réduire le risque de maladies cardiovasculaires. Les β-glucanes sont également utilisés comme agents de texture dans divers produits nutraceutiques et cosmétiques, et comme suppléments de fibres solubles.
Il faut noter que le docteur Samuel Abraham est actionnaire de GN Corporation, au Japon, qui fabrique le Nichi Glucan.
 How ASOs work in the human body. Image by Larissa Nitschke
How ASOs work in the human body. Image by Larissa Nitschke

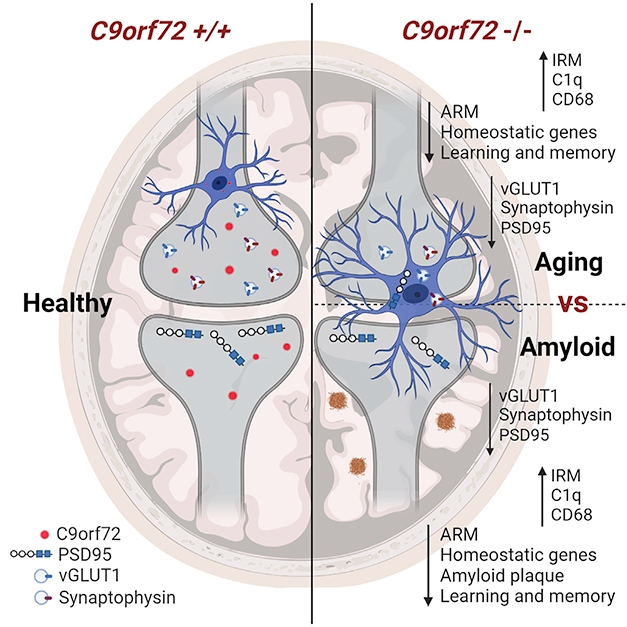

 Cellular stress response is an umbrella concept for molecular changes that cells undergo in response to exposure to extreme temperature, viral infection, toxins, stroke or injury.
Cellular stress response is an umbrella concept for molecular changes that cells undergo in response to exposure to extreme temperature, viral infection, toxins, stroke or injury. 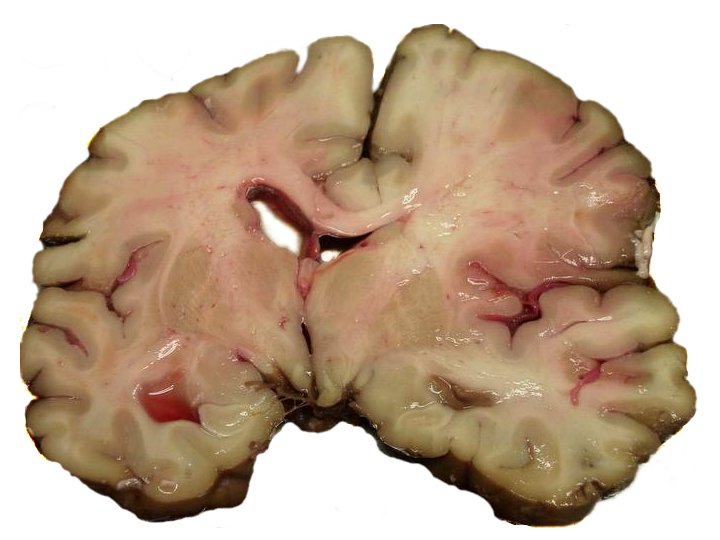 A slice of brain of a person who had a stroke
By Marvin 101 - via Wikipedia
A slice of brain of a person who had a stroke
By Marvin 101 - via Wikipedia