Several articles have recently appeared that should make the pharmaceutical industry reflect on the value of therapies that suppress human proteins or decrease the expression of human genes.
This is the case, for ASOs, this is also the case for the many proposals for immunotherapies against human proteins. We obviously remember the hundreds of unsuccessful clinical trials against ALS or Parkinson's, and the 2,500 unsuccessful trials against Alzheimer's disease.
How did we get there?
In the past 30 years many neurodegenerative diseases, including Huntington's disease, Alzheimer's disease, Parkinson's disease and amyotrophic lateral sclerosis (ALS), have been correlated with DNA damage, resulting in incorrect RNA sequences and therefore poorly translated proteins.
Although initially scientists postulated a mechanism of loss of function (the mutated protein having lost its function). At the turn of the century they realized that this was not a plausible explanation, so they expressed the hypothesis of a gain of function. Which is much more difficult to refute. But this hypothesis is attractive because a gain of function mechanism can be easily suppressed with ASO or immunotherapy.
Case of a vaccine against the Tau protein
For example, the results of a phase II trial of AADvac - a vaccine against pathologic forms of the tau protein - were published on June 14 in Nature Aging by Petr Novak and colleagues.
This vaccine met its primary endpoint in this Phase II study, so it appears reasonably appears safe. It also elicited antibody responses in almost all of the participants, who were diagnosed with mild Alzheimer's disease, and attenuated a gradual increase in plasma NfL over the two-year trial.
By analyzing cerebrospinal fluid samples taken from a small subset of volunteers, the scientists were able to confirm that the vaccine reduced the concentration of Tau protein in the cerebrospinal fluid.
Unfortunately, as usual, the vaccine did not slow the cognitive decline of the patients.
Case of an ASO against C9orf72
Antisense oligonucleotide therapy are strands of RNA that prevent protein translation of certain strands of messenger RNA by binding to them, in a process called hybridization.
Researchers led by Rita Sattler of the Barrow Neurological Institute in Phoenix and Robert Baloh of Cedars-Sinai Medical Center in Los Angeles carried out experiments to suppress the C9orf72 gene on a laboratory mouse model that reproduces the disease Alzheimer's disease, or at least the appearance of amyloid plaques.
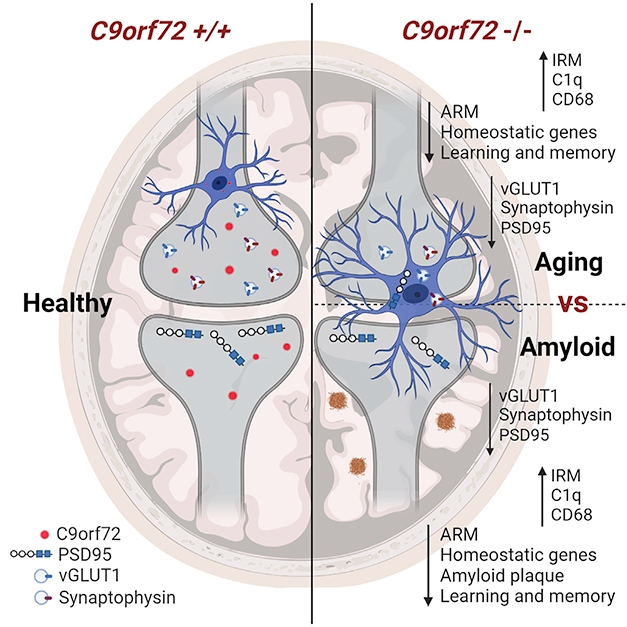
Baloh and his colleagues previously reported in 2016 that in myeloid cells from C9orf72 knockout mice and C9 carriers with ALS / FTD, reporting of lysosomal function leads to escalation of interferon and increased inflammation.
To determine how C9orf72 deficiency affects the way microglia processes plaques and synapses, the researchers made an Alzheimer mice model deficient in C9orf72 expression.
In 3-month-old mice, which begin to develop amyloid plaques, scientists did not detected any obvious effect of C9orf72 deficiency on plaque deposition.
However, at 6 months, the mice lacking C9orf72 had fewer plaques, and those that remained were smaller and more compact than the aggregates of the control mice. Approximately twice as many microglia gathered around each plaque as in the control mice.
At the end of the experiment, the C9orf72 knockout mice had fewer plaques, these were smaller, but neurons had fewer synapses due to overzealous pruning performed by the microglia.
Conclusion
One can only wonder at the interest in therapies that suppress human proteins or decrease the expression of human genes. By what conceptual leap, or by what blindness are we led to think that a potentially deleterious mechanism can become a successful therapy?
