Pour quiconque étudie la recherche contre les maladies neurodégénératives, il est frappant de constater le grand nombre d’études qui affirment chacune avoir identifié un élément clé différent des autres, et qui serait un facteur causatif de la maladie. De plus nombre d’études sont contradictoires.
En épidémiologie, la randomisation mendélienne est une méthode d’utilisation de la variation mesurée des gènes, connue pour exprimer l’effet causal d’une exposition à une maladie dans des études observationnelles, sans avoir besoin d’effectuer un essai clinique randomisé traditionnel. Mieux encore elle permet d’échapper à des biais traditionnels des études épidémiologiques, comme la causalité inverse et la confusion.
Cette méthode a été proposée pour la première fois en 1986 par Gray et Wheatley.
Étant donné que les génotypes sont attribués au hasard lorsqu’ils sont transmis des parents à la progéniture pendant la méiose, la distribution du génotype de la population ne devrait pas être liée aux facteurs de confusion qui affectent généralement les études épidémiologiques d’observation. À cet égard, la randomisation mendélienne peut être considérée comme un essai contrôlé randomisé.
Parce que le polymorphisme est l’instrument, la randomisation mendélienne dépend des études d’association génétique antérieures ayant fourni de bons gènes candidats pour la réponse à l’exposition au risque.
Chacune de ces variantes génétiques sélectionnées doit satisfaire à trois conditions, pertinence, indépendance, restriction d’exclusion.
Parmi les divers facteurs génétiques et environnementaux qui ont été identifiés comme étant associés à la SLA, l’association entre les métabolites des lipides sanguins et la SLA a récemment attiré une attention considérable. Les associations entre les lipides et la SLA sont fortes et comparables en force à de nombreux facteurs de risque de SLA précédemment identifiés.
Les patients SLA souffrent d’une augmentation de la dépense énergétique au repos et d’une perte de poids. Des études observationnelles antérieures ont montré que les patients SLA souffrent fréquemment de dyslipidémie. La dyslipidémie est caractérisée par des niveaux anormaux de lipoprotéines de haute densité (HDL), de lipoprotéines de basse densité (LDL), de cholestérol total (TC) et de triglycérides (TG).
L’association positive entre la dyslipidémie et la SLA suggère que des taux élevés de lipides non HDL peuvent jouer un rôle protecteur dans la progression de la SLA. Conformément aux études observationnelles sur l’homme, des recherches avec des modèles de souris SLA ont également montré que la survie globale des souris SLA est réduite sous la restriction calorique. Cependant, la relation entre la dyslipidémie et la SLA est également controversée, des résultats contradictoires ont été rapportés pour les taux de lipides sériques basaux, la cause de la dyslipidémie et la relation entre les taux de lipides sériques et la progression de la maladie SLA.
Par exemple, de nombreuses études observationnelles de suivi de la SLA n’ont observé aucune association entre la dyslipidémie et la SLA. De plus, certaines études ont montré que les patients atteints de SLA souffrent souvent d’hypolipidémie - qui est principalement caractérisée par de faibles niveaux de LDL - chez les hommes et les femmes SLA. L’association entre l’hypolipidémie et la SLA est en outre confirmée dans un modèle de SLA de souris. Les résultats contradictoires sur la relation entre les niveaux de lipides et la SLA peuvent être dus en partie à la taille relativement petite des échantillons utilisés dans les études précédentes et en partie à des facteurs de confusion non contrôlés qui sont inévitables dans les études d’observation.
Il est difficile de déterminer l’impact causal des lipides sur la SLA au moyen d’études d’essais contrôlés randomisés traditionnels, car ces études nécessitent nécessairement un suivi à long terme, sont coûteuses et souvent contraires à l’éthique. Par conséquent, il est souhaitable de déterminer la relation causale entre les lipides et la SLA par des études observationnelles. La randomisation mendélienne est un puissant outil statistique pour examiner la relation causale et estimer les effets causaux dans les études d’observation.
Des scientifiques ont étudié les effets causaux de quatre traits lipidiques sanguins sur le risque de SLA:
- la lipoprotéine haute densité,
- la lipoprotéine basse densité (LDL),
- le cholestérol total,
- et les triglycérides.
Les auteurs ont d’abord sélectionné les SNP (variantes génétiques) qui peuvent servir de variables instrumentales valides pour chacun des quatre traits lipidiques (HDL, LDL, TC et TG).
En tirant parti des variables de l’instrument à partir de plusieurs études d’association à grande échelle sur le génome dans les populations européennes et asiatiques, les auteurs ont effectué l’une des analyses de randomisation mendélienne les plus importantes et les plus complètes réalisées à ce jour sur la relation causale entre les lipides et la SLA. Parmi les quatre lipides, ils ont constaté que seul le LDL est causalement associé à la SLA et qu’un niveau plus élevé de LDL augmente le risque de SLA dans les populations européennes et est-asiatiques.
La grande taille de l’échantillon utilisée dans cette étude permet aux auteurs d’établir pleinement un effet causal positif du facteur modifiable LDL sur la SLA dans les populations européennes et d’Asie de l’Est. La relation causale inférée entre le LDL et la SLA est robuste en ce qui concerne le choix des méthodes statistiques et est soigneusement validée par diverses analyses de sensibilité.
L’effet causal positif du LDL sur la SLA suggère que le développement futur de stratégies pour réduire les niveaux de LDL réduirait probablement la charge de morbidité de la SLA. Le LDL est un facteur de risque modifiable dont les niveaux peuvent être réduits grâce à diverses stratégies d’intervention. Par exemple, des changements alimentaires tels qu’une augmentation de la consommation de fibres, une augmentation de la consommation de phytostérol et une augmentation de la consommation de noix peuvent tous conduire à une réduction des niveaux de LDL.
Des restrictions de l’apport alimentaire en cholestérol, des restrictions dans les régimes riches en glucides et des restrictions dans la consommation d’acides gras trans peuvent également réduire les niveaux de LDL. Outre le mode de vie et les changements alimentaires, la réduction du LDL peut être obtenue par une thérapie médicamenteuse.
Il ne s'agit pas d'une avancée concernant le mécanisme d'apparition de la maladie, mais cela introduit un outil de gestion de celle-ci.
L’élaboration future de stratégies de réduction des LDL et l’élaboration de politiques publiques pour promouvoir de telles stratégies entraîneraient probablement une réduction de la charge de morbidité de la SLA dans la société.
Publicité

Ce livre retrace les principales réalisations de la recherche sur la SLA au cours des 30 dernières années. Il présente les médicaments en cours d’essai clinique ainsi que les recherches en cours sur les futurs traitements susceptibles d’ici quelques années, d’arrêter la maladie et de fournir un traitement complet en une décennie ou deux.



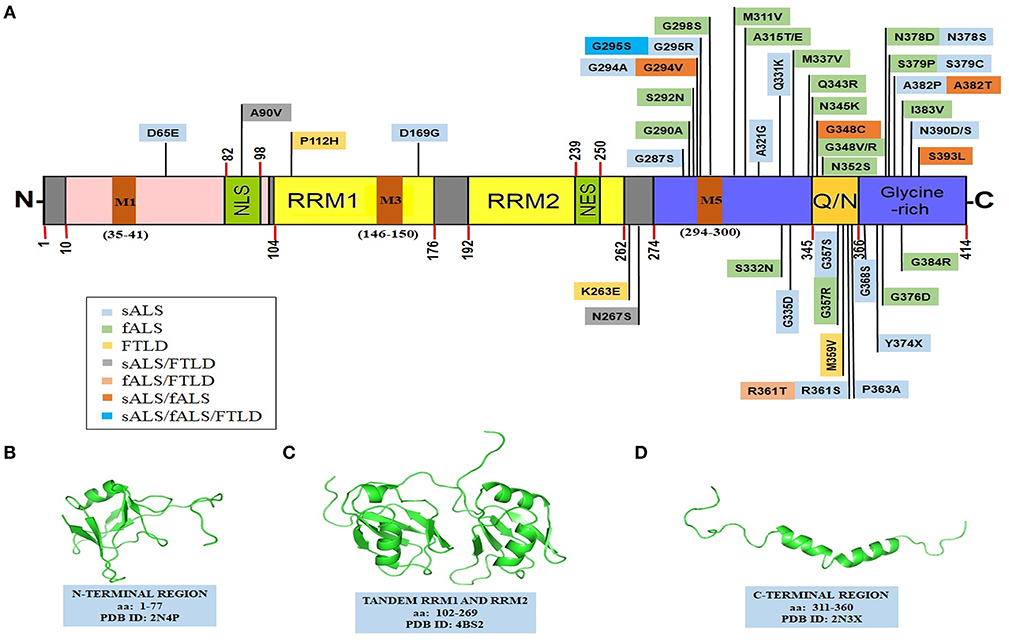 Les auteurs ont montré qu'en présence d’une acidification, même très faible, le TDP-43tRRM se replie complètement et s'oligomérise pour former une «forme β» riche en feuillets β. La forme β a une structure ordonnée et stable qui ressemble aux fibrilles amyloïdes que l'on trouve dans la maladie d'Alzheimer!
Les auteurs ont montré qu'en présence d’une acidification, même très faible, le TDP-43tRRM se replie complètement et s'oligomérise pour former une «forme β» riche en feuillets β. La forme β a une structure ordonnée et stable qui ressemble aux fibrilles amyloïdes que l'on trouve dans la maladie d'Alzheimer! La nature exacte des problèmes apparaît cependant être variable dans ces différentes maladies; par exemple les concentrations de ganglioside
sont réduites dans la maladie de Parkinson et la maladie de Huntington, mais augmentées dans la maladie d’Alzheimer et y a des altérations dans les deux directions pour la SLA.
La nature exacte des problèmes apparaît cependant être variable dans ces différentes maladies; par exemple les concentrations de ganglioside
sont réduites dans la maladie de Parkinson et la maladie de Huntington, mais augmentées dans la maladie d’Alzheimer et y a des altérations dans les deux directions pour la SLA.