Le syndrome de soins post-intensifs (PICS) décrit un ensemble de troubles qui sont courants chez les patients ayant subis une maladie grave et/ou des soins intensifs. Étant donné que la majorité de la littérature en médecine des soins intensifs se concentre sur les résultats à court terme (par exemple, la survie), la compréhension de l’évolution du malade sur le long terme, est relativement limitée, puisque celui-ci est alors considéré comme étant guéri.
Les troubles cognitifs comprennent des déficits de mémoire, d'attention, de vitesse de traitement mental et de résolution de problèmes. Ces déficiences touchent jusqu'à 80% des personnes ayant éprouvé une maladie grave. Les symptômes de la plupart des patients s'améliorent voire disparaissent complètement au cours de la première année qui suit le traitement en unité de soins intensifs.
La physiopathologie sous-jacente de la déficience cognitive chez les survivants des soins intensifs n'est pas bien comprise, mais une inflammation prolongée peut jouer un rôle important
Il a été montré sur des animaux de laboratoire, que High Mobility Group Box 1 (HMGB1), une protéine libérée dans les lésions tissulaires et au cours d'une inflammation sévère, subsiste à une concentration élevée longtemps après le traumatisme et peut provoquer une inflammation hippocampique et des troubles cognitifs.
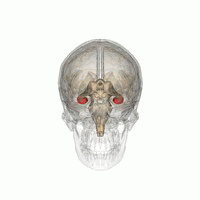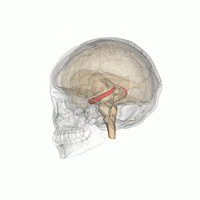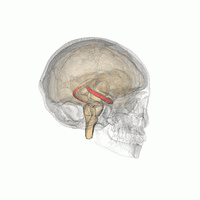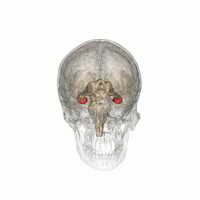 Source: Life Science Databases(LSDB) via Wikipedia.
Source: Life Science Databases(LSDB) via Wikipedia.
Les humains et les autres mammifères ont deux hippocampes, un de chaque côté du cerveau. L'hippocampe fait partie du système limbique. Dans la maladie d'Alzheimer, l'hippocampe est l'une des premières régions du cerveau à subir des dommages; la perte de mémoire à court terme et la désorientation font partie des premiers symptômes. Les personnes atteintes de lésions hippocampiques bilatérales étendues peuvent souffrir d'amnésie antérograde: c'est à dire l'incapacité de former et de conserver de nouveaux souvenirs.
La forme de plasticité neurale connue sous le nom de potentialisation à long terme (LTP) a été initialement découverte dans l'hippocampe et a souvent été étudiée dans cette structure. La LTP est considérée comme l'un des principaux mécanismes neuronaux par lesquels les souvenirs sont stockés dans le cerveau.
Un traitement anti-HMGB1 administré plusieurs jours après une maladie grave peut atténuer le déclin cognitif chez la souris
Des chercheurs du Karolinska Institutet en Suède ont mené une étude de suivi prospective sur 6 mois des taux plasmatiques de HMGB1 et de la fonction cognitive chez les survivants des soins intensifs (essai clinique NCT02914756). 917 patients admis aux soins intensifs ont été dépistés, parmi ceux-ci 100 patients ont été inclus dans l’essai clinique, et ils ont été soumis à des tests de la fonction cognitive et à la mesure des taux plasmatiques de HMGB1 à 3 et 6 mois après la sortie
Les observations ont été effectuées chez ces patients montrent une élévation significative du plasma HMGB1 à 3 et 6 mois après la sortie, et est associée à un dysfonctionnement cognitif.
La source cellulaire de ce HMGB1 systémique est inconnue, mais il est à noter que le HMGB1 est habituellement sécrété par les cellules immunitaires (comme les macrophages, les monocytes et les cellules dendritiques) en tant que médiateur des cytokines de l'inflammation.
Compte tenu de ces propriétés pro-inflammatoires bien établies du HMGB1 extracellulaire, cela suggère une inflammation continue sans résolution de l’inflammation. À la lumière des résultats expérimentaux sur l'atténuation du dysfonctionnement cognitif chez des animaux de laboratoire par la thérapie anti-HMGB1, il est tentant de se demander si le blocage de l'activité pro-inflammatoire du HMGB1 chez les survivants en USI pourrait améliorer les résultats cognitifs.