A new article in the journal BioSocieties, published by Drs. Shlomo Guzmen-Carmeli and David A. Rier, from the Department of Sociology and Anthropology at Bar-Ilan University, tell the story of CliniCrowd, an Israeli company established to test the effectiveness of nutritional supplements like mannitol, cinnamon or cherries in Parkinson's or Alzheimer's disease.
CliniCrowd's model emphasizes speed, efficiency and creativity in dealing with a particular kind of unfinished science, involving potential orphan drugs which, of natural origin, cannot be patented.
Indeed, scientific questions do not all have the same chances of being explored by officially accredited scientists.
The term “unfinished science” refers in sociology to areas of research identified by societal movements as having potentially important social impacts which are however not funded, or incomplete or even completely ignored. This research, although often initiated, is ultimately not carried out for financial, theoretical, ideological or even political reasons.
The classic model of Parsons (1951) of the patient's role as patient supposes that all the action lies with the doctor (the expert), who acts on the passive patients who themselves remain passive, because a priori incompetent.
However, this evolved towards the end of the 20th century, in particular with the fight against AIDS. In 1987, the Community Based Research Initiative, a partnership of physicians and community patients, began a pivotal clinical trial of a treatment for pneumocystis pneumonia, then the main threat to AIDS patients.
The trial provided important clinical data, quickly influenced clinical practice, and was even used by the United States Food and Drug Administration (FDA) in the approval process.
Activists have also formed groups to identify and obtain (sometimes, via smuggling) potential treatment not available in the United States. They criticized drug companies for their high prices and inability to study a wider range of compounds. They particularly attacked the FDA's reliance on very slow and expensive randomized clinical trials (the traditional “gold standard”).
In 2004, the PatientsLikeMe community site was started by two brothers and a friend of a patient with amyotrophic lateral sclerosis (ALS). It opened in 2006 as an online platform, allowing patients with ALS to share their downloaded and anonymized clinical data, assess their own progress, exchange advice and support. , and to contribute more generally to emerging clinical knowledge on the disease.
Finally, PatientsLikeMe is currently a for-profit company, as it was acquired in 2019 by a large healthcare management company. They sell aggregated and anonymized data to academic and professional customers such as pharmaceutical and medical device companies.
In Israel, defense elites and high-tech start-ups are often considered some of the brightest and most innovative in society, trained to think creatively, collaborate and take risks.
Dan Vesely, is a retired Israeli general and high tech entrepreneur. Here's how he describes his response to his terrifying Parkinson's diagnosis in 2013: "If there’s a problem, deal with it. No crying over spilled milk or grieving about my misfortune, about what I ‘won’ [said cynically]. Come on, what do we do next? We think of solutions. [interview, January 24, 2018]"
Vesely, obviously dissatisfied with the treatment options available to him, then turned to acquaintances for help. A small group of entrepreneurs have gathered around him to research the published research on Parkinson's disease.
They quickly noticed the published - and forgotten - study on the possible effect of mannitol on patients with Parkinson's disease. Vesely and some partners contacted Professor Dan Segal of Tel Aviv University, who had co-led the research team, and asked to meet with him:
"It had not yet been tested on humans. So I made an appointment .... Prof. Segal told us his story, described the experiment, and said it's all simply been shelved, there's no incentive for the pharmaceutical companies. at each other and said, 'So we'll take it!' The professor said, 'Who exactly are you? You brash Israelis, who are you?' But it was clear to us that if you can't go through the door , you go through the window. [Vesely interview, Jan. 24, 2018]"
The heartbreaking story of the abandonment of the study born affected the group of friends. Vesely then resolved to test the mannitol on himself. However, its partners dissuaded it from being ineffective for the community because it was totally inconclusive. Instead, together they decided to test mannitol on a number of patients with Parkinson's disease.
In the absence of the support of a pharmaceutical company ready to invest in clinical research, they then sought to test mannitol as if it were a military operation.
They adopted a model, marrying patient self-experimentation with crowdsourcing techniques. Inspired by similar crowdsourcing projects like PatientsLikeMe, the group then planned to create a website for patients with Parkinson's disease who would agree to take mannitol regularly for an extended period.
This alternative is not, however, a real substitute for “* classic *” clinical trials. The survey platform would indeed lack a control group and patient monitoring would be carried out on site on a voluntary and independent basis, and not by a doctor. Nevertheless, this survey platform would generate preliminary data to justify the need for more formal clinical research which would be a result of great value in itself.
The founders of CliniCrowd initially considered marketing mannitol directly, but decided not to, to avoid conflicts with their research. But the founders of CliniCrowd nonetheless chose to register it as a company rather than a non-profit organization. This reflected their primary motivation to “get the job done” as quickly and efficiently as possible, through entrepreneurial tactics, rather than adopting the identity and tactics of social activism. In addition, Israeli non-profit organizations are more regulated than commercial companies.
So they created the company in August 2016. They recruited qualified staff with experience in planning and conducting clinical trials to create the company's platform, and then started recruiting patients using patient forums and media exposure.
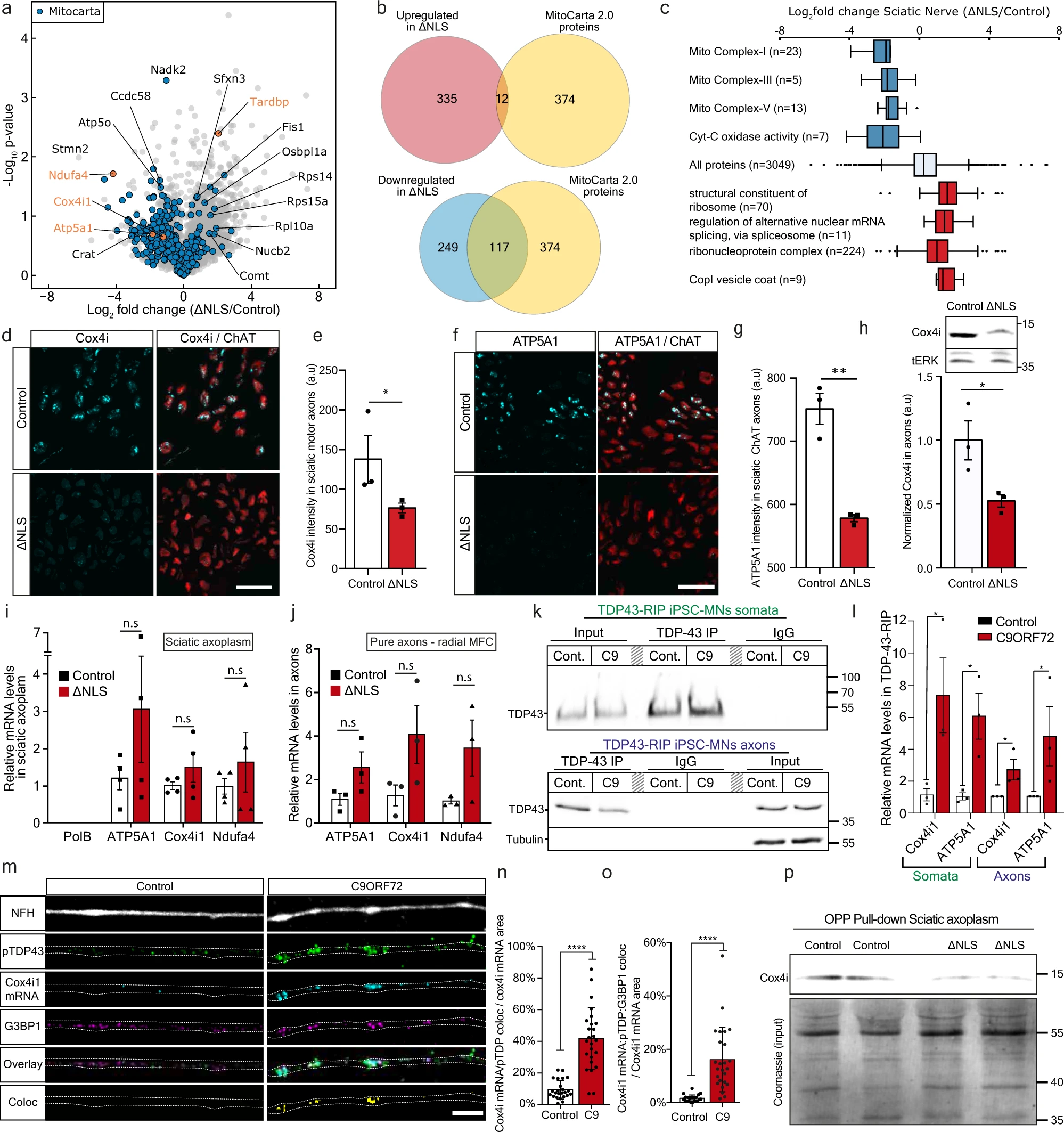
At the start of 2021, 2,480 patients had registered on the platform dedicated to the research of mannitol for Parkinson's disease. Of these, 1,364 (55%) had completed questionnaires more than once. The platform allows patients to record and track data related to their disease and (while maintaining anonymity) compare this data with that of other members of the community. It is also possible to share the stored data with the attending physician.
CliniCrowd's efforts have unfolded in several stages. As Parkinson's patients on the platform began taking mannitol and regularly filling out questionnaires about their symptoms, the next step was to attract accredited scientists to conduct larger trials.
CliniCrowd's initial data helped generate public pressure, which in turn led to a formal clinical study, launched in 2018 at the Hadassah Medical Center in Jerusalem. This study (https://clinicaltrials.gov/ct2/show/NCT03823638), conducted with public funding, examines the effects of mannitol on Parkinson's disease. As of June 2021, the study was continuing, but had slowed down somewhat due to the coronavirus and its severe impact on the medical system. Additional studies, at universities and medical centers in the UK and US, are expected to begin shortly. As far as the authors of this article are aware, at the time of writing, however, these are limited studies.
Nonetheless, there has already been a major shift in the way scientists view mannitol research. As Vesely, the patient-founder of CliniCrowd, noted:
"It gives me great satisfaction that the studies we are currently talking about [the clinical research underway in Jerusalem and expected further studies] would not have taken place, nor would they have received funding or the attention of the medical establishment and the public, without the buzz and especially the clinical indications that CliniCrowd achieved in the wake of the surveys. [interview, July 7, 2019"
Researchers involved in planning the clinical trial confirmed in interviews with the authors that without public pressure, it is unlikely that a trial would have been initiated.
In fact, CliniCrowd's position vis-à-vis the biomedical establishment has evolved over the course of its short history. In early interviews, founders sharply criticized the pharmaceutical industry. For example, in the first interview with CEO Amir Sadeh, he describes the decision to start the business as follows:
"The goal is to create something that cannot be ignored and make available to the public what the pharmaceutical companies are trying to hide from us. Because they [such 'ignored' compounds] do not generate income, they do not make a profit, so it's better not to know about them at all. But now we're exposing them, showing their nakedness in public, telling them it's inexpensive and accessible. It treats the cause rather than the symptoms, and that's why it's the worst thing for the pharmaceutical companies to find a solution to Parkinson's disease. Ten million people, five billion dollars a year — as far as they're concerned, let's just treat the symptoms. It's cynical but that's the way it is…. [T] he benefit of the patients is not the paramount interest of the companies or the doctors, because they are waiting for the next seminar in the Seychelles, courtesy of one company or another. [interview December 3, 2017]"
Yet this initial position of "rebels against the pharmaceutical industry" was created by elite members of the Israeli establishment,
With this approach, CliniCrowd obviously found it difficult to gain the trust and support of the medical establishment.
From interviews the authors conducted with patients who started taking mannitol between 2016 and 2018, it appears that those who saw their doctors have encountered substantial resistance to adopting mannitol as a remedy. The doctors' objections included comments such as, "this is a good woman's medicine" and "you had better get a rabbi's blessing."
At a conference of neurologists in early 2017, CliniCrowd delegates had only a few minutes to present their action, and most conference attendees ignored their speech. Such contempt is reminiscent of the opposition to the community production of knowledge about AIDS more than a generation ago.
The interviews clearly showed that the choice to adopt terms such as "dietary supplements" and "functional foods" reflects CliniCrowd's tactical decision to redefine mannitol as a new substance in the food supplement market.
Here's how CEO Sadeh described the change, in a follow-up interview: "We started out thinking we would call the venture Ampha, as opposed to Pharma. But the more we got into it, the more we realized that was not the point. Like Netflix doesn’t mean all movie theatres are closed, and Airbnb hasn’t replaced hotels, and Uber hasn’t replaced taxis, so CliniCrowd won’t replace the pharmaceutical companies. We fill a void and add something extra. If we started out by setting ourselves against the pharmaceutical companies, now we’re not against them, we’ll be in favour. We’ll complement them. Let’s shift the playing field. Instead of acting on the fiery and aggressive pharmaceutical playing field, let’s move the field elsewhere....And as long as the whole world of medicine doesn’t dance according to the interests of the pharmaceutical companies, we’ve done something great. [interview July 7, 2019]"
The rebranding of mannitol as a functional food proved to be a valuable maneuver, enabling CliniCrowd. This has helped promote acceptance of mannitol among physicians and patients.
Indeed, in the second half of 2018, the authors observed a change in attitude among doctors. Three doctors interviewed for the study told us that once they realized it was a dietary supplement, they stopped protesting: “It’s a dietary supplement. It may not help, but it is not harmful”.
As one neurologist explains: "I think no doctor likes it when the patient comes and says, 'Listen, I've found a treatment.' Most of the time I have to make sure his feet are on the ground, and I must explain why, most probably , in his case it won't work. This was also my initial response to mannitol, complete resistance, not wanting them to take it .... The attitude changes when there is already information and a mass of patients who have collated and documented its use in an orderly manner. Moreover, they didn't come and say this is a magic drug, but rather that it may help with some of the symptoms .... I suggest to patients, especially at the beginning, that they should read about mannitol. I definitely don't exclude it, in fact quite the opposite."
To understand how the patients themselves experienced this, consider Menachem [pseudonym], 68, diagnosed four years earlier. Asked about the experience of taking mannitol and participating in the online questionnaire, he replied: "My participation in the experiment has turned my world around. I come to the doctor and update him, see? I, Menachem, taught the neurologist that there is such a thing as mannitol, and that I am taking part in an experiment with other patients. When I go to see him, he immediately stands up! "Welcome", he says, "tell me how you are getting on". There is a sense that we are colleagues, and that I am doing something incredibly important. There is something in [mannitol] that helps, it’s not a magical cure, or maybe I no longer suffer. But there is an improvement in my sleep, my sense of smell, and also my difficulty in movement. [interview Oct. 30, 2019]"
Note the ease Menachem describes in his relationship with the doctor, his feeling of being an expert, his delight and agency he feels about being involved. These are all so important to him that he mentions them even before his improved health, which he attributes to taking mannitol on a regular basis.
In conclusion, CliniCrowd demonstrated a new way of approaching "* unfinished science *", using participatory research to generate public pressure and influence with which to formally attract scientists to test low potential compounds. profit. CliniCrowd represents an intersection of scientific knowledge, technologies, practices, it is also the product of a sustained process of dissemination and decentralization of expertise.