La sclérose latérale amyotrophique (SLA), souvent appelée maladie de Charcot dans le monde Francophone et maladie de Lou Gehrig aux USA, est une maladie mortelle, gravement débilitante et à évolution rapide, affectant les motoneurones du cerveau, du tronc cérébral et de la moelle épinière. Malheureusement, il existe pas de traitements efficaces pour l'ensemble des malades, il reste donc un besoin crucial de trouver de nouvelles interventions capables d'atténuer ses effets.
Bien que l’étiologie de la SLA reste floue, le vieillissement constitue le principal facteur de risque. Le vieillissement est un processus progressif marqué par le déclin fonctionnel d’un organisme au cours de sa vie. Cependant, on ignore encore dans quelle mesure le vieillissement favorise le risque de SLA. Au niveau moléculaire et cellulaire, il existe des caractéristiques spécifiques du vieillissement normal. Ces caractéristiques sont étroitement liées et se chevauchent largement.
De plus, bien que le vieillissement soit un processus normal, il existe des similitudes frappantes au niveau moléculaire entre ces facteurs et la neurodégénérescence dans la SLA.
Neuf caractéristiques du vieillissement ont été initialement proposées :
* instabilité génomique,
* perte de télomères,
* sénescence,
* modifications épigénétiques,
* détection des nutriments dérégulée,
* perte de protéostase,
* dysfonctionnement mitochondrial,
* épuisement des cellules souches
* altération de la communication intercellulaire.
Cependant, ceux-ci ont été récemment (2023) élargis pour inclure la dérégulation de l’autophagie, l’inflammation et la dysbiose. Par conséquent, compte tenu des dernières mises à jour de ces caractéristiques et de leur association étroite avec les processus pathologiques de la SLA, un nouvel examen de leur relation avec la physiopathologie est justifié. Dans cette revue, nous décrivons les mécanismes possibles par lesquels le vieillissement normal a un impact sur les mécanismes neurodégénératifs impliqués dans la SLA, ainsi que les nouvelles interventions thérapeutiques qui peuvent en découler.
La sclérose latérale amyotrophique (SLA) est une maladie mortelle et à évolution rapide qui affecte les motoneurones du cerveau, du tronc cérébral et de la moelle épinière, entraînant une paralysie musculaire progressive. Avec un tel mauvais pronostic et des symptômes gravement débilitants, il est important d’identifier les mécanismes sous-jacents qui déclenchent la SLA. L'âge moyen du diagnostic de la SLA est de 55 ans et le vieillissement est son principal facteur de risque.
Le vieillissement est un déclin lentement progressif et continu du fonctionnement normal d’un organisme au cours du déroulement de sa vie. Il est également marqué par une sensibilité accrue aux maladies et un risque accru de décès.
Il est important de noter que l'Organisation mondiale de la santé (OMS) estime que la proportion de la population mondiale de plus de 60 ans, doublera presque (de 12 % à 22 %) entre 2015 et 2050, ce qui implique que l'incidence des maladies neurodégénératives liées à l'âge telles que la SLA augmentera considérablement dans les décennies à venir. Il est toutefois important de noter que ces estimations pourraient être révisées à l’avenir en raison des circonstances uniques et des défis posés par la pandémie de COVID-19.
La décennie actuelle est pour les Nations Unies celle du vieillissement en bonne santé (2021-2030), une collaboration mondiale dirigée par l’OMS, reconnait l’importance du vieillissement sur la santé.
Il est également essentiel de noter la différence entre la durée de vie (nombre total d'années pendant lesquelles un individu survit depuis sa naissance jusqu'à sa mort) et la durée de vie (nombre total d'années pendant lesquelles un individu reste en bonne santé, sans maladie chronique). Ainsi, vieillir en bonne santé devrait également prendre en compte la durée de vie ainsi que la durée de vie.
Il est bien établi que des changements dans la morphologie et la fonction du cerveau sont présents au cours du vieillissement, impliquant des diminutions de poids et de volume, perte de matière blanche et grise et dégénérescence des neurites et des synapses [1].
Alors que l'effet du vieillissement normal sur la moelle épinière reste peu étudié en comparaison, des altérations significatives y ont été décrites, notamment la perte des motoneurones alpha (α-MN)[2], qui rappelle la SLA. Les cellules musculaires, comme les motoneurones, présentent également bon nombre des caractéristiques classiques du vieillissement. Bien que le vieillissement lui-même soit un processus normal, il existe des similitudes frappantes entre la neurodégénérescence et le vieillissement normal au niveau moléculaire et cellulaire, car les « caractéristiques » spécifiques associées au vieillissement [3] chevauchent de manière significative les mécanismes physiopathologiques impliqués dans la SLA (Fig. 1).
Cependant, la manière exacte dont le vieillissement favorise l’augmentation du risque de SLA reste mal définie. Les caractéristiques moléculaires et cellulaires du vieillissement ont été récemment mises à jour [3]. D'où un nouveau l'examen de la relation entre le vieillissement et la neurodégénérescence dans la physiopathologie de la SLA est justifié et fait l'objet de cette revue.
Fig.1. Caractéristiques moléculaires du vieillissement dans la SLA. Les caractéristiques moléculaires du vieillissement désignent un ensemble de caractéristiques moléculaires et cellulaires interconnectées qui sont largement liées au processus de vieillissement dans divers tissus et organismes. Ces mécanismes offrent un cadre pour comprendre les subtilités du vieillissement. Il est frappant de constater que les caractéristiques moléculaires du vieillissement présentent des similitudes notables et se chevauchent considérablement avec les mécanismes physiopathologiques décrits dans la SLA
Vieillissement et neurodégénérescence
Le vieillissement se produit avec le temps dans tous les organismes, bien qu'il progresse à des rythmes différents au sein d'une espèce. Les différences entre les individus sont dues aux variations de la constitution génétique, de l'environnement, du mode de vie et de l'adaptation [
4,
5] et sont évidents au niveau de l'organisme, des organes, des cellules et des molécules. Nous fournissons ci-dessous un bref aperçu des principales théories du vieillissement.
Théories du vieillissement
Alors que les facteurs qui contrôlent la durée de vie humaine restent flous, les théories actuelles du vieillissement reposent principalement sur en deux grandes catégories.
* La « théorie du décès programmé » propose que le vieillissement normal suive un calendrier biologique (similaire à celui régulant la croissance de l'enfant) qui entraîne des changements dans l'expression des gènes impliqués dans la maintenance cellulaire [
6].
* En revanche, la théorie des « dommages ou erreurs » suggère que le vieillissement est le résultat de dommages progressifs aux cellules et aux organes au fil du temps [
6].
Cependant, il n’existe actuellement aucun consensus sur les causes du vieillissement chez l’homme. De plus, de nombreux mécanismes cellulaires impliqués dans le vieillissement interagissent largement et peuvent donc agir ensemble pour accélérer les processus moléculaires sous-jacents.
La théorie programmée du vieillissement
La théorie programmée du vieillissement peut être divisée en trois sous-types.
* La théorie de la « longévité programmée » implique que le vieillissement résulte de changements dans l'expression des gènes, conduisant à des déficits associés à l'âge et à un phénotype cellulaire sénescent ultérieur [
7].
* Deuxièmement, la « théorie endocrinienne » propose que les hormones agissent comme des horloges biologiques pour contrôler le taux de vieillissement [
8].
* Troisièmement, la « théorie immunologique » affirme que la fonction du système immunitaire est à son apogée pendant la puberté, mais qu'elle décline par la suite, ce qui entraîne une susceptibilité accrue à l'inflammation [
9].
Théorie des dommages ou des erreurs du vieillissement
La théorie des « dommages ou erreurs » du vieillissement peut être divisée en cinq sous-types.
* Premièrement, la théorie de « l'usure » propose que les composants cellulaires s'usent naturellement avec le temps en raison d'une utilisation répétée et constante [
6].
* Deuxièmement, la théorie du « taux de vie » affirme que la durée de vie d'un organisme devient plus courte avec des taux de consommation basale d'oxygène plus élevés [
10,
11].
* Troisièmement, la « théorie des liens croisés » [
12 ] propose que les protéines deviennent réticulées puis s'agrègent au fil du temps [
12,
13] .
* La théorie des « radicaux libres » [
14,
15] suggère que le superoxyde et d'autres radicaux libres s'accumulent et endommagent les composants cellulaires (acides nucléiques, lipides, sucres et protéines) au cours du vieillissement [
15]. Bien que les antioxydants neutralisent cela dans une certaine mesure, cela finit par devenir inefficace au cours du vieillissement normal [
15]
* Enfin, la théorie des « dommages somatiques à l'ADN » propose que les dommages à l'ADN se produisent continuellement dans les cellules. Alors que ces lésions sont initialement réparées, l'augmentation des dommages au fil du temps entraîne des mutations qui altèrent l'intégrité du génome et donc la fonction cellulaire. Des dommages à l'ADN nucléaire et mitochondrial sont impliqués dans ce processus [
16].
Génétique et vieillissement dans la SLA
On ne sait pas exactement comment le vieillissement augmente le risque de SLA, mais cela implique probablement une combinaison de facteurs génétiques, environnementaux et liés à l'âge [
17]. Alors que la plupart (~ 90 %) des cas de SLA surviennent de manière sporadique, la proportion restante est familiale et peut fournir un aperçu de la physiopathologie sous-jacente [
17]. Les expansions répétées d'hexanucléotides (GGGGCC) dans le premier intron du gène du cadre de lecture ouvert 72 du chromosome 9 (
C9ORF72) sont la cause génétique la plus courante de la SLA familiale (~ 40 %) et sporadique (~ 8-10). %), ainsi que la maladie connexe, la démence frontotemporale (DFT), à la fois sporadique (~ 5 à 10 %) et familiale (~ 25 à 30 %) [
18]. La DFT affecte principalement les lobes frontaux et temporaux du cerveau et les patients présentent une combinaison de symptômes cognitifs, comportementaux et/ou moteurs, bien que ceux-ci puissent varier considérablement [
19]. Certaines personnes atteintes de DFT peuvent également développer des symptômes de SLA et vice versa, appelés « ALS-FTD » [
20].
Il existe donc un chevauchement génétique et pathologique important entre la SLA et la DFT. Trois mécanismes principaux sont impliqués dans la neurodégénérescence induite par les mutations de l'hexanucléotide
C9ORF72 ;
* production d'ARN toxiques,
* traduction non-AUG (RAN) pour produire des protéines répétées dipeptides (DPR)
* haploinsuffisance due au manque de protéine C9ORF72 [
21].
Les mutations dans les gènes codant pour la superoxyde dismutase 1 ( SOD1) et la protéine 43 de liaison à l'ADN TAR (TDP-43) provoquent respectivement ~ 20 % et ~ 4 % de cas de SLA familiale [
22–
24].
* TDP-43 est une protéine de liaison ARN/ADN normalement située principalement dans le noyau. Cependant, la présence de formes pathologiques de TDP-43, impliquant sa troncature, son agrégation anormale et sa mauvaise localisation dans le cytoplasme, sont la marque caractéristique de presque tous (~ 97 %) les cas de SLA [
25].
* Fused in Sarcoma (FUS) est une autre protéine de liaison à l'ARN présentant des similitudes structurelles et fonctionnelles avec le TDP-43 et des mutations dans le FUS provoquent également ~ 4 % des cas [
26].
* Plus de 30 autres gènes ont été associés à la SLA familiale, bien que chacun représente une plus faible proportion de cas. Ces gènes incluent CCNF, CCHHD10, ATXN2, KIF5A, hnRNPA2/B1, UBQLN2, TBK1. OPTN, PRPH, NEK1, VCP, et
PFN1, entre autres [
17,
27,
28].
Alors que la SLA implique la dégénérescence et la mort des motoneurones, les cellules gliales, qui assurent un rôle de soutien important aux neurones, contribuent également à la physiopathologie via des mécanismes autonomes non cellulaires. Les astrocytes régulent le flux sanguin dans le système nerveux central (SNC), recyclent les neurotransmetteurs et forment la barrière hémato-encéphalique. Les microglies sont impliqués dans la phagocytose, la réponse immunitaire, la neuroinflammation, ainsi que dans la surveillance et l'activation immunitaires. Ainsi, ils agissent comme les cellules immunitaires du SNC [
29]. Les oligodendrocytes myélisent les axones neuronaux dans le SNC pour faciliter la transmission synaptique et fournir un soutien métabolique aux neurones, et les cellules de Schwann myélinisent les axones neuronaux dans le système nerveux périphérique (SNP). Ces dernières cellules jouent également un rôle important dans le maintien de la fonction de la jonction neuromusculaire (NMJ) [
30].
Les manifestations cliniques de la SLA sont dues à une perte de la fonction musculaire volontaire, normalement facilitée par les motoneurones. au NMJ [
31,
32]. Auparavant, on pensait que la SLA affectait principalement les motoneurones, et que l’implication des muscles squelettiques était une conséquence secondaire. Cependant, le rôle du muscle dans la pathogenèse de la SLA est de plus en plus reconnu (récemment examiné içi [
33]).
Caractéristiques moléculaires du vieillissement dans la SLA
Les caractéristiques moléculaires et cellulaires du vieillissement sont définies par des critères spécifiques [
3] ;
* (a) un critère doit changer en fonction du temps au cours du processus de vieillissement,
* (b) il doit être renforcé par une accélération expérimentale du vieillissement,
* (c) la modulation du critère doit inhiber, arrêter ou même inverser le vieillissement.
Neuf critères de vieillissement ont été initialement proposés (en 2013) [
3] : instabilité génomique, perte de télomères, sénescence, modifications épigénétiques, détection des nutriments dérégulée, perte de protéostase, dysfonctionnement mitochondrial, épuisement des cellules souches et altération de la communication intercellulaire.
Cependant, ces caractéristiques ont été récemment (2023) [
3] élargies pour inclure la dérégulation de l’autophagie, l’inflammation et la dysbiose.
Il est important de noter cependant que ces douze caractéristiques liées à l'âge se chevauchent de manière significative et sont fortement interdépendantes, avec de nombreuses interactions entre ces voies (Fig.
1). Plusieurs sont impliqués en tant que caractéristiques « primaires » et moteurs du processus de vieillissement [
3], y compris l'instabilité génomique, le dysfonctionnement des télomères, la dérégulation épigénétique et la dérégulation de la protéostase (Fig.
2). En revanche, les caractéristiques « antagonistes » font référence à des réactions cellulaires aux dommages, notamment la détection des nutriments, le dysfonctionnement mitochondrial et la sénescence. Enfin, les caractéristiques « intégratives » reflètent le manque de capacité de la cellule à faire face aux dommages associés à l’âge, impliquant des défauts de communication intercellulaire, un épuisement des cellules souches et une dysbiose. Ci-dessous, nous détaillons chacune des douze caractéristiques et leur lien avec les mécanismes de neurodégénérescence dans la SLA.

Fig. 2
Principaux facteurs du vieillissement et SLA : Le vieillissement et la neurodégénérescence dans la SLA sont des processus complexes influencés par une combinaison de facteurs génétiques, environnementaux et cellulaires. Bien que les causes précises du vieillissement et de la SLA ne soient pas entièrement comprises, l'instabilité génomique, l'attrition des télomères, les altérations épigénétiques, le dysfonctionnement de la protéostasie, l'autophagie dérégulée et le dysfonctionnement mitochondrial sont considérés comme les principaux facteurs.
Principales caractéristiques du vieillissement et leur lien avec la SLA
Instabilité génomique
L'instabilité génomique fait référence à la fréquence élevée de mutations au sein du génome [
34]. Cela peut résulter de sources à la fois exogènes et endogènes, telles que les agents environnementaux et les erreurs de réplication de l'ADN, respectivement. La réponse aux dommages de l'ADN (DDR) fait référence aux voies de signalisation qui détectent et réparent normalement les dommages de l'ADN, et l'efficacité de la réparation de l'ADN diminue avec le vieillissement [
3]. L'instabilité génomique résulte soit d'altérations de l'architecture nucléaire, de dommages à l'ADN nucléaire et/ou mitochondrial et de mécanismes de réparation de l'ADN défectueux [
35]. Cependant, bien que l'instabilité génomique augmente de manière significative avec le vieillissement, il manque des preuves directes montrant qu'elle module spécifiquement le vieillissement.
Altérations de l'architecture nucléaire
L'architecture du noyau maintient de multiples aspects de la stabilité du génome. Cela implique principalement la lame nucléaire, un maillage filamenteux situé sous l’enveloppe nucléaire qui attache les protéines et la chromatine. Les protéines lamines nucléaires sont ses principaux constituants et sont fortement associées au vieillissement et à la stabilité du génome. Il est important de noter que des mutations dans les gènes codant pour plusieurs de ces protéines provoquent des troubles du vieillissement accéléré tels que le syndrome de Hutchinson-Gilford-progeria (HGPS ou progeria) [
36,
37], qui résulte d'une forme tronquée anormale de Lamin A (progérine), qui s'accumule également normalement avec l'âge [
38]. La dérégulation de la lamine B1 perturbe le complexe Shelterin et entraîne une instabilité des télomères dans les cellules humaines [
39].
Les défauts du noyau et l'altération du transport nucléocytoplasmique sont bien décrits dans la SLA. Une pathologie des pores nucléaires est détectée dans le cerveau de patients sporadiques atteints de SLA, TDP-43 et C9ORF72 [
40]. Les formes pathologiques du TDP-43 perturbent l'architecture nucléaire et les complexes de pores nucléaires dans la SLA [
40 ]. Le variant FUSR521G associé à la SLA interagit avec les nucléoporines, qui forment le complexe des pores nucléaires, et perturbe le transport nucléocytoplasmique [
41]. L'ARN C9ORF72 et les DPR interagissent également avec et perturbent divers composants de la machinerie de transport nucléaire tels que les récepteurs de transport nucléaire, la Ran GTPase, les nucléoporines et les protéines de l'enveloppe nucléaire [
42]. Cependant, la morphologie nucléaire n'est pas modifiée dans C9ORF72 ALS/FTD [
43]. La perte de la nucléoporine NUP50 a été impliquée comme facteur de risque de SLA [
44]. Les mutations liées à la perte de la kinase-1 (NEK1) liée à la mitose A (NIMA) dans les motoneurones dérivés de cellules souches pluripotentes induites (CSPi) perturbent également l'architecture nucléaire et l'importation de protéines [
45].
Dommages à l'ADN nucléaire
Les cellules sont très sujettes aux dommages à l'ADN et les insultes surviennent à un rythme de dizaines de milliers. par jour et par cellule [
34]. Les mutations somatiques s'accumulent normalement avec le temps et le taux de formation est inversement corrélé à la durée de vie [
3 ]. Au cours du vieillissement normal, l'efficacité des mécanismes de réparation de l'ADN diminue, entraînant une accumulation de dommages à l'ADN [
35]. En outre, des mutations dans plusieurs protéines de réparation de l’ADN provoquent plusieurs troubles progéroïdes humains, liant directement les déficits de réparation de l’ADN au vieillissement. Les cassures d'ADN double brin (DSB) constituent le type de dommage le plus toxique. Dans les neurones, elles sont réparées principalement par le mécanisme de jonction d'extrémités non homologues (NHEJ), sujet aux erreurs. Les neurones sont également sujets aux dommages oxydatifs de l'ADN, qui sont réparés par réparation par excision de base (BER) [
46]. Il existe désormais de nombreuses preuves de dommages à l'ADN dans la physiopathologie de la SLA. Plusieurs protéines centrales à la SLA, notamment notamment C9ORF72, FUS, TDP-43, SOD1, NEK1, C21orf2, la sénataxine et la protéine 1 contenant de la valosine (VCP), sont connues pour fonctionner dans la réparation de l'ADN [
47]. Nous et d'autres avons montré que le TDP-43 est recruté dans les foyers γH2AX où il fonctionne dans le NHEJ [
48], interagit avec Ku 70 et est impliqué dans la réparation des boucles R [
49,
50]. FUS interagit avec l'histone désacétylase 1 (HDAC1) pour réparer les DSB [
51 ] et il fonctionne également dans le BER en médiant le recrutement dépendant de PARP1 de XRCC1/DNA Ligase IIIα (LigIII). C21orf72 interagit avec NEK1 et serait impliqué dans la réparation du DSB [
52–
54]. Le VCP et la sénataxine sont également impliqués dans le maintien de l'intégrité génomique en facilitant la transcription, la réplication de l'ADN et le DDR [
55,
56].
Les dommages à l'ADN sont également induits par des formes pathologiques des mêmes protéines dans la SLA [
57]. Le mutant TDP-43 de la SLA présente une activité altérée dans NHEJ, ce qui perturbe l'homéostasie de la boucle R et induit une pathologie TDP-43 [
48,
58]. La pathologie TDP-43 est associée à une instabilité du génome, englobant des changements d'épissage, des mutations somatiques et des fusions de gènes [
59]. La perte de TDP-43 dans le noyau est en corrélation avec une accumulation accrue de DSB [
60,
61 ].
De même, le mutant FUSR521C associé à la SLA induit des dommages à l'ADN et des défauts d'épissage de l'ARN [
62]. La perte de FUS nucléaire altère la ligature de l'ADN en inhibant le recrutement de XRCC1/LigIII [
63 ], induisant la formation d'agrégats et la neurodégénérescence [
64]. De plus, des variantes d’autres protéines associées à la SLA induisent également des dommages à l’ADN. Les gènes de réparation de l'ADN sont activés en réponse aux dommages à l'ADN causés par les mutations SOD1G93A dans les motoneurones dérivés des iPSC [
65]. Les mutations d'hexanucléotides dans
C9ORF72 induisent des dommages à l'ADN dans les cellules neuronales et les motoneurones des patients atteints de SLA [
66]. Cela a été associé à des déficiences dans la réparation des boucles DSB et R et dans l'ubiquitylation de H2A [
67]. Les DPR C9ORF72 poly-glycine arginine (poly-GA) et poly-proline-arginine (poly-PA) induisent des DSB et la phosphorylation de l'ataxie télangiectasie mutée (pATM) [
68]. Il existe également des preuves liant les défauts de réparation de l'ADN à la perte des motoneurones.
Ercc1Δ/− souris dépourvues de mécanismes de réparation de l'ADN, réparation par excision de nucléotides (NER), réparation de liaisons croisées inter-brins, NHEJ et recombinaison homologue (HR) affichent une perte aberrante des motoneurones, une activation des microglies et des astrocytes, un dysfonctionnement de l'appareil de Golgi, un stress génotoxique et une pathologie NMJ [
69]. Cependant, ni la pathologie TDP-43 ni la pathologie FUS n'ont été détectées dans les motoneurones de ces souris, ce qui indique que la perte de
Ercc1 à elle seule est suffisante pour induire une pathologie liée à la SLA [
69,
70]. Ensemble, ces données impliquent qu'il existe une forte corrélation entre la SLA et les dommages à l'ADN, ce qui soulève la possibilité que le vieillissement normal augmente l'instabilité génomique et donc le risque de neurodégénérescence. Cependant, cela n'a pas été démontré directement.
Dommages à l'ADN mitochondrial
L'ADN mitochondrial (ADNmt) est particulièrement vulnérable aux mutations somatiques liées à l'âge. s proximité des sites de phosphorylation oxydative et manque de protection par les histones [
71 ]. Il accumule les dommages oxydatifs en fonction de l'âge [
71]. De plus, bien que les mécanismes de réparation de l’ADNmt ne soient pas aussi bien étudiés que ceux de l’ADN nucléaire, ils semblent moins efficaces [
71].
Les mutations de l'ADNm et l'augmentation du stress oxydatif sont impliquées dans les deux phénomènes de vieillissement. et le développement de la SLA [
72,
73]. Le TDP-43 de type sauvage et le TDP-43Q331K mutant se localisent dans les mitochondries et déclenchent la libération d'ADNmt à travers le pore de transition de perméabilité mitochondriale [
74]. L'accumulation d'ADNmt active ensuite la voie cGAS/STING, induisant une neuroinflammation et une neurodégénérescence [
74]. L'ADNmt cytoplasmique est également présent dans la moelle épinière des patients atteints de SLA et dans les motoneurones dérivés des iPSC [
74]. Par conséquent, ensemble, ces études impliquent que des dommages à l'ADNmt sont présents dans la SLA, bien que cela ne soit pas bien caractérisé.
Attrition des télomères
Les télomères sont des séquences d'ADN répétitives non codantes (TTAGGG) trouvées aux extrémités distales des chromosomes qui protègent l'intégrité du génome. lors de la réplication. Au cours du vieillissement normal, la longueur des télomères diminue et les rongeurs ayant des télomères courts ou longs présentent respectivement une inhibition ou une prolongation de la durée de vie [
3]. Le raccourcissement des télomères est donc l'une des caractéristiques majeures du vieillissement impliquée dans de nombreuses maladies liées à l'âge [
75]. La transcriptase inverse de la télomérase (TERT) empêche le raccourcissement des télomères en maintenant la longueur des télomères [
75], et bien que le raccourcissement des télomères induit une instabilité génomique et des dommages à l'ADN, il est reconnu comme une caractéristique distincte du vieillissement [
3].
Une dérégulation de la longueur des télomères a également été décrit dans la SLA. L'inactivation de la télomérase entraîne un raccourcissement des télomères et un phénotype SLA accéléré dans le modèle de souris SOD1G93A [
76]. De plus, un raccourcissement des télomères dépendant de l'âge a été détecté dans les motoneurones iPSC de patients C9ORF72 [
77]. Cependant, une récente étude de séquençage du génome entier a conclu que des télomères plus longs constituent un facteur de risque de SLA et aggravent le pronostic, y compris dans le cerveau [
78]. De même, une longueur de télomère plus longue est associée à la FTD [
79]. Par conséquent, il est possible que le maintien d’une longueur équilibrée des télomères soit essentiel dans la SLA et que les modifications de la longueur des télomères, à la fois l’allongement et le raccourcissement, soient toutes deux pertinentes pour la neurodégénérescence. En revanche, des études d'association à l'échelle du génome n'ont trouvé aucune association entre la longueur des télomères et la SLA dans les leucocytes, ce qui implique que la longueur des télomères est spécifique au type de cellule [
80]. Ainsi, ces résultats contrastés impliquent que davantage d'études sont nécessaires pour caractériser la longueur et l'activité des télomères dans la SLA.
Altérations épigénétiques
L'épigénétique fait référence aux changements héréditaires dans la régulation de l'expression des gènes, indépendamment de la séquence d'ADN. On sait que de multiples modifications épigénétiques se modifient au cours du vieillissement [
81], y compris la méthylation de l'ADN, l'acétylation des histones, le remodelage de la chromatine et la régulation des ARN non codants [
81]. Ces altérations affectent la réplication et la réparation de l'ADN, la transcription et l'inactivation des gènes, la division cellulaire et le maintien de la longueur des télomères [
81].
La méthylation de l'ADN sur la cytosine est l'une des modifications la plus étudiée en ce qui concerne les modifications épigénétiques.
La chromatine, contenant à la fois de l'ADN génomique et des histones, régule l'accessibilité de la machinerie de transcription et donc l'expression des gènes. Au cours du vieillissement, des altérations de la chromatine se produisent, notamment un remodelage structurel et des modifications de l'architecture de la chromatine, une perte d'histones et des modifications post-traductionnelles des histones. L'acétylation des histones est régulée par les histones acétyltransférases (HAT) et les histones désacétylases (HDAC) [
82 ,
83]. Une diminution de l'acétylation globale des histones entraîne une dérégulation de l'expression des gènes métaboliques et une homéostasie métabolique [
82]. L'hyper-ou hypo-acétylation des histones est régulée par l'homéostasie HAT/HDAC, et un déséquilibre dans ce processus induit des défauts dans la réponse intégrée au stress et les mécanismes de réparation de l'ADN [
84]. Les inhibiteurs d'HDAC ont été impliqués comme stratégie thérapeutique pour prévenir le vieillissement [
85 ].
Il existe de plus en plus de d'indications à propos du rôle des modifications épigénétiques dans la pathogenèse de la SLA, en particulier en relation avec l'expansion des répétitions C9ORF72.
86]. La méthylation accrue d'un îlot CpG près de la répétition GGGGCC dans le promoteur C9ORF72 diminue l'expression de la protéine C9ORF72 [
87]. De plus, la méthylation de l'ADN accélérée par l'âge dans l'île CpG 5' est associée à un phénotype de maladie plus grave, à une apparition précoce et à une courte durée de la maladie chez les patients C9ORF72 [
88]. Les histones H3 et H4 subissent une hyper-méthylation du promoteur CpG-island [
89] chez les patients SLA et FTD [
90 –
93]. L'hyperméthylation inhibe également la formation de foyers d'ARN et l'agrégation de DPR dans la SLA [
94]. Moins de méthylation nucléaire de 5-méthylcytosine (5mC) et de 5hmC a été détectée dans les motoneurones inférieurs présentant une pathologie TDP-43 par rapport à ceux dépourvus de pathologie [
95]. De plus, les motoneurones dérivés d'iPSC provenant de variantes
FUS associées à la SLA expriment plus d'ADN méthyltransférases et présentent plus de méthylation dans la région promotrice de
FUS [
96]. Des études menées sur des souris SOD1G93A ont également identifié une méthylation aberrante de l'ADN et de l'ARN (augmentée ou diminuée) dans la moelle épinière et les muscles squelettiques par rapport aux souris témoins [
97].
Des altérations épigénétiques de la chromatine sont également décrites dans la SLA. Un complexe de remodelage de la chromatine, le complexe de facteurs associés au gène 1 neuronal Brahma (Brg1) (nBAF), qui dans les neurones régule la différenciation, l'expansion dendritique et la fonction synaptique, manquait dans les motoneurones en culture exprimant le FUSR521G ou TDP- associé à la SLA. 43G348C [
98]. L'expression du type sauvage TDP-43 perturbe également la dynamique de la chromatine en raison d'un fonctionnement altéré de l'enzyme de remodelage de la chromatine CHD2 chez
Drosphilie [
99]. L'homéostasie HAT/HDAC est altérée dans le cerveau et la moelle épinière des patients FUS-ALS [
84]. L'inhibition de l'HDAC à l'aide de l'ACY-738 rétablit l'acétylation globale des histones, améliore la survie et réduit les anomalies métaboliques dans un modèle murin surexprimant le FUS de type sauvage [
100]. Les inhibiteurs d'HDAC ont été examinés de manière approfondie dans des modèles de SLA (souris SOD1G93A, modèles de souris FUS et C9ORF72 (détaillés plus en détail dans la section "
Interventions thérapeutiques pour le vieillissement et les maladies liées au vieillissement") [
85,
101].
Perte de protéostasie
L'homéostasie des protéines, ou « protéostasie », fait référence au réseau dynamique de processus qui régulent la machinerie de synthèse, de repliement, de trafic et de dégradation des protéines [
102]. La protéostasie dépend du bon fonctionnement des chaperons moléculaires, de l'autophagie, du système du protéasome de l'ubiquitine (UPS) et de la dégradation associée au réticulum endoplasmique (ER) (ERAD). La perte de protéostase se produit si ces mécanismes de contrôle de la qualité des protéines échouent, ce qui peut entraîner l'accumulation de protéines mal repliées ou agrégées [
103]. Au cours du vieillissement normal, l’efficacité de la protéostase diminue et l’accumulation qui en résulte de protéines mal repliées endommagées et agrégées est une caractéristique clé du vieillissement et de la neurodégénérescence. Les agrégats de lipofuscine – des granules composés de protéines et de lipides mal repliés en tant que sous-produit de la digestion lysosomale – s'accumulent également dans les motoneurones au cours du vieillissement normal [
104–
106]. Les protéines peuvent également être modifiées post-traductionnellement au cours du vieillissement par des dommages oxydatifs causés par des espèces réactives de l'oxygène (ROS) ou des sucres, et la modification ultérieure entraîne la formation de produits finaux de glycation avancée (AGE)[
107]. Le taux de traduction des protéines diminue avec l'âge, et un allongement ralenti de la traduction induit un mauvais repliement des protéines et un vieillissement [
108]. L'effondrement de la protéostasie fait référence à la panne ou à la défaillance de la machinerie cellulaire responsable du maintien de l'homéostasie des protéines et est impliqué comme un facteur important du vieillissement cellulaire chez l'homme [
103,
109].
L'expression de protéines chaperons telles que les protéines de choc thermique (HSP ) diminue avec le vieillissement [
110], ce qui implique que Le repliement des protéines devient altéré avec l’âge. L'administration de HSP70 humaine recombinante à des souris retarde la sénescence, améliore l'activité du protéasome et les fonctions cognitives, réduit les taux de lipofuscine cérébrale et prolonge la durée de vie [
111]. Nourrir les jeunes mouches des fruits avec des AGE et de la lipofuscine inhibe l'UPS, ce qui accélère le vieillissement et réduit la durée de vie [
112]. De même, un autre chaperon, l'oxydoréductase protéine disulfure isomérase (PDI), protège contre le vieillissement cellulaire dans plusieurs modèles, notamment les cellules souches mésenchymateuses humaines sénescentes réplicatives (hMSC RS), les hMSC HGPS, les hMSC du syndrome de Werner (WS) et les hMSC primaires humaines [
113]. De plus, la stabilisation de la protéostase dysfonctionnelle à l'aide du chaperon chimique 4-phényl butyrate (PBA) améliore le comportement cognitif et inhibe le vieillissement [
114].
La SLA pathologique se caractérise par des agrégats de protéines mal repliés implique fortement des défauts de protéostase en physiopathologie [
102]. La dérégulation de la plupart des mécanismes de protéostase et de contrôle de la qualité des protéines est également bien décrite dans la SLA, notamment des défauts de l'autophagie, de l'UPS, du transport ER-Golgi et de l'ERAD [
102]. De nombreux chaperons moléculaires sont également dérégulés dans la SLA, notamment les protéines PDI et les HSP [
102]. Les protéines PDI ont également été associées à la SLA en tant que mécanisme de protection et facteur de risque génétique [
115–
118].
La formation de granules de stress (SG) est de plus en plus reconnue dans le maintien de la protéostasie [
119]. Les SG sont des organites cytoplasmiques sans membrane (également appelés condensats biomoléculaires) composés de protéines et d'ARN [
120–
123]. Fonctionnellement, ils sont impliqués dans le stockage des biomolécules et comme emplacements de tri de l'ARNm pour réguler la traduction et la stabilité de l'ARNm [
124,
125]. La formation de SG est régulée par séparation de phase liquide (LLPS), le processus par lequel les protéines et les acides nucléiques en solution se séparent en gouttelettes liquides (semblables aux gouttelettes d'huile qui se forment dans l'eau) [
126]. Les SG s'assemblent et se désassemblent en réponse à des conditions exogènes ou environnementales, favorisant ainsi la survie lors d'un stress cellulaire [
127]. La LLPS est pilotée par des protéines avec des domaines intrinsèquement désordonnés, qui incluent des protéines mal repliées associées à la SLA, y compris le TDP. -43 et FUS [
128]. Des études récentes ont montré que les SG séquestrent les protéines mal repliées, les empêchant de s'accumuler dans le noyau ou le cytoplasme, maintenant ainsi la protéostase [
119].
Cependant, les SG anormaux perturbent la protéostase et, au cours du vieillissement normal, les défauts de régulation du montage/démontage normal et de la dynamique des SG sont liés à la perte de protéostasie [
33].
Les SG sont présents dans les agrégats pathologiques de la SLA. De plus, ils sont impliqués dans la formation d'inclusions protéiques mal repliées via la nucléation de ces agrégats [
129]. Le TDP-43 se localise dans les SG en présence de stress ER, de stress oxydatif, de stress mitochondrial, de stress osmotique et d'inhibition du protéasome [
49,
130–
134]. La variante SOD1G93A associée à la SLA se localise avec les SG, contrairement à la SOD1 de type sauvage [
135,
136] . La colocalisation des agrégats TDP-43 et des marqueurs SG a été détectée dans les tissus de patients SLA [
120,
130 ,
137, ;
138], bien que les études cellulaires n'aient pas pu détecter co-localisation entre le mutant TDP-43 A315T, M337V et les SG dans des conditions de stress [
139,
140 ]. De même, une colocalisation entre le FUS R495X et les SG mutants de la SLA a été rapportée dans des lignées cellulaires, des neurones primaires et des tissus humains [
137,
141–
145].
Macroautophagie dérégulée
L'autophagie est un processus catabolique responsable de la dégradation et du recyclage des composants cellulaires. La macroautophagie est le principal type d'autophagie, qui implique la formation de vésicules à double membrane, ou autophagosomes. La dérégulation de la macroautophagie est bien décrite dans le vieillissement et a récemment été classée comme une caractéristique distincte de la protéostase, car les organites et les composants cellulaires non protéiques sont également sujets à la macroautophagie [
3]. On sait que l'expression des gènes liés à l'autophagie, notamment
ATG5, ATG7 et
OPTN, diminue avec l'âge [
146,
147]. Cela entraîne l'accumulation d'agrégats de protéines et d'organites dysfonctionnels au cours du vieillissement [
146]. De plus, la stimulation ou l’activation de l’autophagie augmente la durée de vie et la durée de vie des individus. les humains et les organismes modèles [
146]. L'autophagie est également réduite dans les échantillons musculaires obtenus auprès de patients âgés [
148]. L'élimination du gène 7 lié à l'autophagie (
ATG7) chez la souris entraîne une atrophie musculaire accrue, une inflammation musculaire, une structure anormale et une durée de vie réduite [
148].
La macroautophagie dérégulée est impliqué dans la neurodégénérescence dans la SLA [
149] et la SLA -variantes associées dans
C9ORF72, SOD1, TARDBP, TBK1, FUS, FIG4, OPTN, UBLN2, SQSTM1, CHMP2B, ALS2 dérégulent la macroautophagie [
150]. Lorsque l'autophagie est inhibée génétiquement ou pharmacologiquement, le vieillissement est accéléré et la toxicité des motoneurones est accrue dans la SLA [
146,
151]. L'autophagie joue également un rôle essentiel dans l'élimination des agrégats de protéines associés à la neurodégénérescence dans la SLA [
152]. Une activation accrue des protéines de l'autophagie est détectée chez les souris transgéniques SOD1G93A [
153 ]. De même, la progestérone est neuroprotectrice grâce à l'activation de l'autophagie chez les souris SOD1G93A [
154]. C9ORF72 lui-même interagit avec le complexe Rab1a et Unc-51-like kinase 1 (ULK1) pour initier l'autophagie via la formation d'autophagosomes [
155] et la perte de C9ORF72 altère l'autophagie [
156,
157]. Les DPR C9ORF72 co-localisent avec l'inclusion d'inclusions p62-positives, ce qui suggère que les DPR sont ciblés pour élimination par l'UPS et/ou l'autophagie [
158]. La mutation TDP-43A315T active le stress du RE et induit l'autophagie pour éliminer les agrégats de protéines mal repliés [
159 ].
Caractéristiques antagonistes du vieillissement dans la SLA
Sénescence cellulaire
La sénescence est impliquée comme une caractéristique importante et un moteur du processus de vieillissement. De nombreuses études ont montré que la sénescence régule les phénotypes associés à l'âge et est présente dans les maladies liées à l'âge [
160,
161]. Les cellules sénescentes étaient auparavant considérées comme nocives car leur élimination prolonge la durée de vie des souris [
162]. Cependant, des études plus récentes sur le foie ont montré que les cellules sénescentes ont un impact positif sur le vieillissement en bonne santé et la durée de vie et pourraient jouer un rôle fonctionnel important dans le vieillissement [
163]. Dans les cellules en cycle, la sénescence est caractérisée par un état d'arrêt éternel du cycle cellulaire bien qu'elles restent métaboliquement actives [
164]. Les dommages à l'ADN dans le noyau (principalement sous forme de DSB) et le raccourcissement des télomères sont des caractéristiques clés de la sénescence [
165–
169]. Le DDR associé à la sénescence implique ATR, ATM et p53, qui induit l'activation des inhibiteurs de kinases dépendant de la cycline p16, p21 et p27 et une hyperphosphorylation de la protéine du rétinoblastome (Rb), qui entraîne le retrait du cycle cellulaire [
170].
La sénescence est induite par divers stimuli endogènes et exogènes, notamment le stress oxydatif, la neuroinflammation, l'activation oncogène, l'inactivation des gènes suppresseurs de tumeurs et le dysfonctionnement mitochondrial [
168,
171]. Pendant la sénescence , les cellules subissent plusieurs modifications phénotypiques, notamment de profondes modifications de la chromatine et du sécrétome, ainsi que l'activation du suppresseur de tumeur [
172]. Le phénotype sécrétoire associé à la sénescence (SASP) est une caractéristique importante de la sénescence qui induit une inflammation via l'accumulation de cytokines pro-inflammatoires, de chimiokines et de facteurs de croissance [
173]. Par conséquent, les cellules sénescentes peuvent induire des altérations significatives du microenvironnement cellulaire via SASP, ce qui peut aggraver l'inflammation [
174]. On pense que les microglies de la substance blanche sont le principal type de cellules subissant une sénescence dans le SNC au cours du vieillissement [
175].
Les neurones étant post-mitotiques, ils ne subissent pas de sénescence « réplicative » classique, on pensait donc à l’origine que ce mécanisme était limité aux cellules en division. Cependant, les neurones expriment des marqueurs de sénescence, la SASP est présente dans le cerveau vieillissant et des découvertes récentes ont révélé que les neurones subissent un processus similaire à la sénescence en réponse au stress (« sénescence prématurée induite par le stress ») [
170,
176–
179]. Par conséquent, la sénescence des neurones vieillissant normalement peut compromettre leur viabilité et augmenter leur vulnérabilité à des agressions supplémentaires [
180]. Cependant, notre compréhension de la sénescence des neurones reste limitée [
181].
La sénescence a été décrite dans la SLA, bien qu'elle ait été détectée principalement dans les cellules gliales. Dans la moelle épinière lombaire de rats SOD1G93A symptomatiques, des microglies présentant des caractéristiques de sénescence ont été détectées [
174]. Des marqueurs de sénescence, notamment la perte de l'expression de la lamine nucléaire B1 et une augmentation significative de p16INK4a, p53, de la métalloprotéinase-1 matricielle (MMP-1) étaient présents par rapport aux rats transgéniques non transgéniques ou asymptomatiques [
174]. Il est intéressant de noter que d’autres types de cellules de la moelle épinière lombaire dégénérative, notamment les motoneurones ChAT-positifs et les astrocytes exprimant GFAP, présentaient également une coloration nucléaire p16INK4a. De même, dans les astrocytes générés à partir d'iPSC d'individus atteints de SLA sporadique et de patients ALS-C9ORF72, il y a eu une augmentation significative de l'expression des marqueurs de sénescence [
182]. Le cerveau des patients atteints de SLA présente également un nombre élevé d'astrocytes sénescents [
183 ].
Les cellules satellites sont des cellules souches adultes des muscles squelettiques qui résident entre les fibres musculaires et les membranes basales et qui s'auto-répliquent et/ou se différencient en nouvelle forme de nouvelles fibres musculaires suite à une blessure [
184] . Une sénescence a été rapportée dans ces cellules chez des souris gériatriques, entraînant un arrêt de la régénération des fibres musculaires [
185]. L'inactivation de l'homologue de la région 1 d'insertion Mo-MLV du lymphome B (Bmi1) entraîne des phénotypes de type sénescence dans les jeunes cellules satellites [
185]. Protéine arginine méthyltransférase 7 (PRMT7) [
186] est associée à la capacité de régénération musculaire et son expression diminue en fonction de l'âge [
187]. Une diminution de la masse musculaire squelettique, une altération de la régénération des cellules satellites et une sénescence prématurée ont été détectées chez des souris knock-out PRMT7 [
186]. Ensemble, ces résultats soutiennent l'idée selon laquelle la sénescence joue un rôle dans le développement de la SLA, bien que cela ne soit pas bien caractérisé [
182].
Dysfonctionnement mitochondrial
Les mitochondries sont des organelles multifonctionnelles associées depuis longtemps au vieillissement. Ils constituent les principales sources d'énergie de la cellule, et régulent également l'immunité innée, l'inflammation et l'apoptose [
188 ]. Au cours du vieillissement, les fonctions mitochondriales sont altérées par des défauts du potentiel membranaire, une diminution de la capacité respiratoire, une production accrue de radicaux libres, une réduction du renouvellement et de la dynamique, ainsi que par l'accumulation de mutations dans l'ADNmt [
73,
188].
Le dysfonctionnement mitochondrial est largement décrit dans la SLA [
73,
188,
189]. Des déficiences du complexe 1 de la chaîne respiratoire mitochondriale sont présentes dans les motoneurones obtenus à partir de coupes de moelle épinière lombaire de patients SLA sporadiques [
106,
190 ]. Une diminution du potentiel de membrane mitochondriale est présente dans les motoneurones humains dérivés de C9ORF72 et du mutant TDP-43M337V dérivés de CSPi [
191] et fibroblastes [
192]. L'haploinsuffisance de C9ORF72 altère la bioénergétique et la fonction des mitochondries, ainsi que l'expression des complexes de chaînes de transport d'électrons [
193,
194]. La surexpression des DPR C9ORF72 (en particulier poly-GR) induit des dommages à l'ADN mitochondrial, perturbe le potentiel de la membrane mitochondriale et augmente la production de ROS [
195]. Poly-GR se lie à l'ATP synthase mitochondriale Atp5a1, induisant des défauts dans la structure et la morphologie des mitochondries [
196]. Une accumulation anormale de mitochondries est présente dans les motoneurones de la moelle épinière des souris transgéniques mutantes TDP-43A315T et SOD1G93A [
197] et un dysfonctionnement des mitochondries et des anomalies de transport sont présents dans les cellules exprimant le mutant SLA TDP-43Q331K, M337V [
198–
200] et mutant SOD1G93A, G85R [
201]. Mutations dans UBQLN2P497S [
202] et FUSR514G induisent également des anomalies mitochondriales [
203]. La SOD1 oxydée liée à la SLA déclenche un dysfonctionnement mitochondrial et une sénescence cellulaire, ce qui accélère encore le vieillissement, établissant ainsi un lien plus direct entre le stress oxydatif, la SLA et le vieillissement[
204]. Ensemble, ces résultats suggèrent que le dysfonctionnement mitochondrial est étroitement associé aux principales protéines pathologiques de la SLA.
Détection des nutriments dérégulée
Au cours du vieillissement, on observe un déclin des principales voies de signalisation métabolique liées au vieillissement et à la neurodégénérescence [210], impliquant les facteurs de croissance adrénergiques, dopaminergiques, insuline/insuline-like. 1 (IGF1), protéine kinase activée par l'AMP (AMPK), voies sirtuine (SIRT) et mTOR. L'IGF-1 est le principal médiateur de l'action de l'hormone de croissance (GH) qui module le métabolisme des glucides via l'insuline. Le vieillissement entraîne une réduction des taux d'IGF-1 et de GH [
205], y compris dans le cerveau [
206 ]. L'AMPK est un capteur de l'état énergétique cellulaire et son activation rétablit l'équilibre énergétique. De plus, une activité réduite de l'AMPK est impliquée dans le vieillissement [
207]. mTOR, une sérine-thréonine protéine kinase, est un régulateur négatif du vieillissement qui favorise la SASP [
208]. Chez la levure, les vers et les mouches, le blocage de mTORC1 prolonge la durée de vie [
209 ].
Le nicotinamide adénine dinucléotide (NAD +) est un coenzyme central au métabolisme énergétique et un cofacteur essentiel dans les réactions redox cellulaires et les activités SIRT [
211]. Il influence la réparation de l'ADN, le remodelage de la chromatine et la sénescence, et des niveaux réduits de NAD + sont détectés au cours du vieillissement [
212,
213]. Les SIRT sont une famille de sept protéines qui régulent la survie et le métabolisme des cellules/tissus et possèdent de nombreuses fonctions associées au vieillissement. Cela inclut la réparation de l'ADN et la stabilité du génome, la sénescence et la fonction mitochondriale, et ils inhibent le stress oxydatif, l'inflammation et l'apoptose [
214,
215]. SIRT-1, 2, 3 et 6 augmentent également la durée de vie d'espèces allant des mouches des fruits aux mammifères [
216–
218 ]. L'expression de SIRT1 diminue avec le vieillissement, donc une expression élevée des SIRT peut protéger contre les événements liés à l'âge [
219].
Les changements dans les voies métaboliques sont associés à l'hétérogénéité et aux diverses caractéristiques cliniques de la SLA. L'inhibition de mTOR chez les souris transgéniques mutantes SOD1G93A accélère la progression de la maladie et augmente la dégénérescence des motoneurones [
220 ]. Cependant, l'inhibition de mTOR est protectrice dans un modèle de souris transgénique impliquant une surexpression de TDP-43 de type sauvage spécifique aux neurones [
221]. La surexpression de l'IGF-1 dans les motoneurones primaires protège contre la toxicité induite par le glutamate dans la SLA [
222]. L'activation de l'AMPK a été détectée dans les motoneurones de patients atteints de SLA ainsi que dans la moelle épinière de souris SOD1G93A [
223]. Une dérégulation du SIRT a été décrite dans la SLA [
224 –
226] et acétylation de la lysine-136 sensible à SIRT1 entraîne le LLPS et l'agrégation pathologique du TDP-43 [
227,
228]. L'activation de SIRT-1 a été étudiée thérapeutiquement à l'aide du resvératrol, qui a initialement montré des effets prometteurs en améliorant la déficience motrice et en prolongeant la durée de vie chez les souris SOD1G93A [
229]. Cependant, il a échoué lors des essais cliniques [
230] .
Critères intégratifs et ALS
Communication intercellulaire altérée
Les cellules peuvent communiquer entre elles soit par des interactions physiques directes, soit par des intermédiaires tels que des vésicules extracellulaires (VE) qui agissent comme messagers intercellulaires. Au cours du vieillissement normal, la qualité de la communication entre les cellules diminue progressivement, ce qui a un impact sur plusieurs processus liés à la SLA. Ceux-ci sont abordés dans les sections ci-dessous.
Sénescence et communication intercellulaire
Les cellules sénescentes sont métaboliquement actives et peuvent communiquer avec les cellules voisines et influencer leur comportement via la signalisation paracrine [
231]. La sénescence est également un élément important de la communication intercellulaire et du vieillissement [
232] via SASP [
233 ]. En plus de la sécrétion de molécules pro-inflammatoires, les cellules sénescentes communiquent également avec d'autres cellules via des ponts intercellulaires liés à la membrane ou des « nanotubes tunnels », qui facilitent les connexions physiques directes entre les cellules [
234]. Le rôle de la sénescence dans le vieillissement et la SLA est décrit dans le
Section caractéristiques intégratives et SLA.
Neuroinflammation et inter- communication cellulaire entre les cellules gliales et les neurones
Au sein du SNC, les neurones, les astrocytes, les microglies et les oligodendrocytes doivent normalement communiquer entre eux et avec l'environnement pour maintenir homéostasie. La santé et la viabilité des motoneurones reposent sur une communication efficace avec les cellules gliales et les muscles squelettiques [
235,
236].
La SLA est une maladie non autonome cellulaire et une communication intercellulaire extrinsèque entre les motoneurones, les microglies, les oligodendrocytes et les astrocytes. est impliqué dans la physiopathologie. Cela se produit par des altérations du soutien des facteurs trophiques aux motoneurones, des facteurs de signalisation qui ont un impact sur les récepteurs des cellules gliales et des changements dans les interactions directes de cellule à cellule [
236]. L'administration intrathécale de LCR provenant de patients atteints de SLA chez des souris réduit l'expression des facteurs trophiques BDNF, du facteur de croissance des fibroblastes 2 (FGF2) et de l'IGF-1 [
237]. Les cytokines pro-inflammatoires et le TNF-α et le ligand Fas (FASL) déclenchant l'apoptose produits par les microglies et les astrocytes activés induisent des dommages aux motoneurones [
238,
239]. Les neurones corticaux de souris traités avec des astrocytes dérivés d'iPSC provenant de patients C9ORF72 présentent une augmentation du stress oxydatif et de la neurotoxicité [
182]. Les oligodendrocytes dégénératifs et morphologiquement altérés sont considérablement augmentés chez les souris mutantes SOD1G93A et sont entourés de microglies activées en cluster [
240]. Les astrocytes dérivés de la SLA familiale post-mortem (SOD 1A4V) et du cerveau des patients atteints de SLA sporadique sont toxiques pour les motoneurones, mais cela est atténué en réduisant l'expression de la
SOD1 dans les astrocytes [
241]. Dans le modèle de souris mutantes SOD1G93A, les astrocytes sénescents présentent moins de soutien aux motoneurones. De plus, les niveaux d'IL-6 augmentent dans les astrocytes des modèles de rongeurs SOD1G93A qui recrutent des cellules immunitaires pour éliminer les cellules sénescentes [
242,
243].
Vésicules extracellulaires et communication intercellulaire
Les EV sont de minuscules structures liées à une membrane, dont la taille varie généralement de 50 à 1 000 nm [
244]. Ils contiennent à la fois des protéines et de l'acide nucléique et sont libérés par divers types de cellules dans des conditions physiologiques et pathologiques [
244]. Les ARN extracellulaires (exARN) sont d'importants médiateurs de communication de cellule à cellule qui sont sécrétés sous forme d'EV ou dans un complexe avec des protéines de liaison à l'ARN (RBP)[
245]. Les véhicules électriques associés à la sénescence sont impliqués dans le DDR et le SASP [
246,
247]. Les niveaux d'EV changent au cours de la sénescence et du vieillissement, bien que leur augmentation ou leur diminution soit controversée [
248–
250].
Les exARN et les véhicules électriques sont également impliqués dans la pathogenèse de la SLA. Certains exARN, notamment l'ARNm, le microARN et l'ARN circulaire, sont présents dans les exosomes et, comme ils sont dérégulés dans la SLA, ils ont été proposés comme biomarqueurs potentiels [
174,
175] [
251 ] (la dérégulation de l'ARN dans la SLA est examinée plus en détail dans le '
Défauts dans le dysfonctionnement de l'ARN section). Des souris transgéniques SOD1G93A libèrent des astrocytes-dEV dérivés contenant SOD1G93A mutant qui se transfèrent aux neurones spinaux et déclenchent sélectivement la mort [
252]. Les microvésicules isolées de patients SLA contiennent des niveaux plus élevés de protéines pathologiques (SOD1, TDP-43, FUS) par rapport aux témoins, contrairement aux exosomes, bien que la taille moyenne des deux types d'EV soit plus grande dans la SLA que celle des témoins [
253].
On sait que les protéines mal repliées se transmettent entre les cellules de la SLA et d'autres maladies neurodégénératives, en particulier la SOD1 [
254,
255]. Plusieurs études ont décrit les caractéristiques « de type prion » de la SOD1 mal repliée, notamment sa capacité à se transférer entre les cellules et à provoquer le mauvais repliement de la SOD1 de type sauvage dans les cellules [
256] et in vivo [
257]. La transmission d'agrégats toxiques via les véhicules électriques n'est pas bien comprise [
244 ]. La SOD1 mal repliée, qu'elle soit de type sauvage ou les variantes associées à la SLA A4V, G93A, G127X, sont sécrétées sous forme d'EV dans les cellules NSC-34 et HEK [
254]. Les astrocytes et les neurones constituent les principales sources d'EV in vivo contenant de la SOD1 mal repliée dans la moelle épinière des souris transgéniques SOD1G93A et des patients SOD1-ALS [
258]. De même, un comportement « de type prion » pour le TDP-43 a été décrit chez la souris [
259,
260]. D'autres études ont démontré que des mécanismes à la fois dépendants et indépendants des exosomes sont impliqués dans la transmission intercellulaire du TDP-43 [
261]. De même, les DPR C9ORF72, poly-GA, poly-GP, poly-GR et poly-PA se transmettent de cellule à cellule par des mécanismes dépendants et indépendants des exosomes [
262].
Neuroinflammation et vieillissement
L'inflammation augmente considérablement au cours du vieillissement normal, tant au niveau systémique que dans le système nerveux (neuroinflammation). Les cellules sénescentes contribuent également à l'environnement inflammatoire persistant via SASP [
162], et leur accumulation entraîne une inflammation soutenue [
263 ]. Les inflammasomes, complexes protéiques multimériques qui activent la caspase inflammatoire 1, font partie intégrante du système immunitaire inné qui s'active au cours du processus de vieillissement [
264]. Cela inclut l'inflammasome de la protéine 3 du récepteur de type nucléotide oligomérisation (NOD) (NLRP3) [
265].
La neuroinflammation joue un rôle important dans le vieillissement du SNC et les conditions pathologiques associées [
266]. Au cours du vieillissement, les microglies et les astrocytes activés présentent des morphologies altérées et produisent des cytokines pro-inflammatoires, conduisant à une neuroinflammation [
267–
269 ]. Lorsqu’ils sont activés, les astrocytes peuvent présenter des phénotypes neurotoxiques, pro-inflammatoires (A1) ou neuroprotecteurs et anti-inflammatoires (A2). De même, les microglies présentent à la fois des états inflammatoires et anti-inflammatoires, respectivement M1 et M2. Séquençage de l'ARN des astrocytes dérivés du cerveau tout au long de la vie des souris [
270 ,
271] révélé régulation positive des gènes du phénotype A1 associés à la neuroinflammation [
271], reliant les astrocytes aux troubles cognitifs au cours du vieillissement. Les souris sont protégées de l'astrogliose réactive liée à l'âge en l'absence de cytokines pro-inflammatoires microgliales, ce qui suggère que les microglies sont responsables de l'initiation du système neuronal. inflammation qui survient avec le vieillissement [
271]. Cependant, les astrocytes exercent des effets néfastes sur les microglies au cours du vieillissement, altérant leurs capacités phagocytaires, entraînant un état pro-inflammatoire prolongé [
272].
La neuroinflammation est bien décrite dans la SLA humaine et dans les modèles animaux [
273]. L'infiltration de lymphocytes périphériques, de cellules tueuses naturelles (NK) et de macrophages, ainsi que l'activation des astrocytes et des microglies et la production excessive de cytokines inflammatoires, sont présentes chez les humains et les souris [
274]. Il est intéressant de noter que l'analyse transcriptomique de la moelle épinière de souris SOD1G93A a révélé un chevauchement significatif (transcriptions partagées à 90 %) entre les modèles d'expression génique associés au vieillissement normal et à la SLA, en particulier l'inflammation et l'activation du système immunitaire [
219]. L'inflammasome NLRP3, ainsi que l'expression de la caspase-1, de l'IL-1β, de l'IL-18 et de NFκB, sont augmentés chez le rat transgénique SOD1G93A [
275]. Dans les astrocytes de la moelle épinière de souris SOD1G93A [
276] et chez les patients SLA sporadiques, des taux élevés de NLRP3, une protéine de type speck associée à l'apoptose contenant un domaine de recrutement de la caspase-1 (ASC), de l'IL18 et de la caspase 1 active, sont présents [
277]. Les microglies chez les souris SOD1G93A et TDP-43Q331K SALS mutantes et de type sauvage expriment NLPR3, ce qui correspond à une expression élevée des composants de l'inflammasome in vivo [
265]. Le TDP-43 se lie aux récepteurs CD14 des microglies, des macrophages et des monocytes, activant le NFκB et stimulant l'inflammasome NLRP3 [
278]. Chez les rats SOD1G93A, la progression de la paralysie était liée à la neuroinflammation et à la toxicité des motoneurones via la microglie [
174]. Il existe des preuves des effets neuroprotecteurs et neurotoxiques des astrocytes et des microglies dans la SLA (examinés dans Clarke et al. 2020) [
279,
280].
Épuisement des cellules souches
Les cellules souches ont des capacités d'auto-renouvellement et de multi-différenciation et régénèrent ainsi la croissance des tissus au cours du vieillissement. Les cellules souches neurales (NSC) sont responsables de la production de neurones pendant le développement prénatal et du maintien du système nerveux tout au long de la vie adulte [
281,
282 ]. Cependant, au cours du vieillissement, la fonctionnalité et la capacité de régénération des NSC se détériorent. Cet épuisement des cellules souches peut être induit expérimentalement par une régulation positive des dommages à l'ADN, une altération des mécanismes de réparation de l'ADN, une diminution de la capacité de régénération, des altérations épigénétiques, une instabilité génomique accrue, une altération de l'homéostasie des protéines, des mitochondries dysfonctionnelles et la sénescence [
290]. Plusieurs études ont identifié des moyens possibles d'améliorer la fonction des cellules souches au cours du vieillissement, par exemple en augmentant les niveaux des facteurs de transcription FOXO4, HSP70 [
290], ou bien en exposant du sang jeune à des animaux âgés par parabiose hétérochronique [
288].
Une méta-analyse de onze études ont démontré que l'isolement et la transplantation de NSC du SNC dans la moelle épinière de souris transgéniques mutantes SOD1G93A ralentissaient la progression de la maladie [
281]. Cela était lié à l'amélioration de la production de facteurs neurotrophiques, à la réduction de la neuroinflammation et à la préservation de la fonction neuromusculaire [
281]. La régénération et le renouvellement des cellules souches âgées peuvent être bénéfiques sur le plan thérapeutique dans les maladies neurodégénératives, notamment la SLA, bien que cela n'ait pas été bien étudié.
Dysbiose
Il est désormais reconnu que le microbiome intestinal joue un rôle essentiel dans la santé et bien-être [
3], y compris le vieillissement [
4,
5], et il est façonné par la génétique, l'âge, le stress, la maladie, les médicaments, l’alimentation et l’environnement. Cependant, le microbiome est dérégulé dans de nombreuses conditions pathologiques, ce que l'on appelle la « dysbiose » [
3]. La plupart des micro-organismes intestinaux sont des bactéries impliquées dans le métabolisme, la défense contre les agents pathogènes, le développement du système immunitaire et la synthèse des vitamines, des acides gras à chaîne courte et d'autres métabolites [
3]. Il est important de noter que le microbiome interagit avec le SNC via l'axe intestin-cerveau, le réseau bidirectionnel reliant le système nerveux entérique au SNC [
2].
Le microbiome intestinal s'établit pendant l'enfance. Bien qu'il présente une diversité significative entre les individus, [
3 ] au cours du vieillissement normal, des modifications dans la composition du microbiote intestinal et une diversité réduite des espèces sont associées à une fragilité, à des fonctions cognitives, à des symptômes dépressifs et à des processus inflammatoires [
3]. De plus, des modèles murins de progeria et de patients atteints de progeria atteints de HGPS ou du syndrome de Nestor-Guillermo progeria (NGPS) présentent une dysbiose, caractérisée par la perte et le gain d'espèces spécifiques [
36]. La transplantation de microbiote fécal entre des souris sauvages et des souris progeria confirme l'existence d'un lien étroit avec la durée de vie/santé [
37,
38] et dans le maintien de la santé cérébrale et de l'immunité pendant le vieillissement [
11]. De même, l'administration de métabolites du microbiote intestinal améliore les pathologies liées à l'âge chez la souris [
2,
3 ]. Collectivement, ces résultats suggèrent que le vieillissement est étroitement associé à la dysbiose.
La dysbiose est également liée à la neurodégénérescence dans la SLA. La dérégulation du microbiome intestinal est en corrélation avec la gravité de la maladie chez les souris transgéniques mutantes SOD1G93A et chez les patients humains [
283]. Le butyrate de sodium est un métabolite bactérien produit dans l'intestin par
Butyrivibrio fibrisolvens, et des niveaux réduits de cet organisme ont été détectés chez la souris SOD1G93A [
284]. Une perméabilité intestinale accrue aux toxines a également été détectée [
284 ] et le traitement des souris SOD1G93A avec du butyrate a également retardé la perte de poids et amélioré la survie [
285. ], ce qui implique que les interventions visant à restaurer le microbiome intestinal peuvent prolonger la durée de vie et la durée de santé dans la SLA. Des altérations du microbiote intestinal ont également été détectées chez des souris mutantes C9ORF72 [
286], et le C9ORF72 lui-même s'est avéré inhiber les réponses inflammatoires systémiques et neuronales induites par les bactéries intestinales [
286]. Ensemble, ces études impliquent que le microbiome intestinal contribue à la fois au vieillissement et à la pathogenèse de la SLA.
Défauts dans les fonctions de l'ARN
Les défauts du métabolisme de l'ARN ne sont pas considérés comme une caractéristique du vieillissement [
3], mais il a été proposé qu'ils soient désignés comme tel, étant donné les preuves croissantes soulignant leur importance pour le vieillissement [
287]. Étant donné que les cellules vieillissantes perdent leur capacité à maintenir le métabolisme de l'ARN [
288] et un métabolisme dysfonctionnement de l'ARN est fortement impliqué dans la physiopathologie de la SLA [
289 ], nous considérons ici cela comme une caractéristique du vieillissement qui est discutée en relation avec la SLA.
Le milieu d'ARN au sein d'une cellule est constitué d'ARN messagers codants ( ARNm) et les ARN non codants (ARNnc), qui interagissent tous deux avec les protéines de liaison à l'ARN (RBP) au sein des complexes ribonucléoprotéiques (RNP). Les RBP jouent des rôles importants dans le métabolisme de l'ARN, notamment l'épissage alternatif du pré-ARNm, le transport et la stabilité, qui sont affinés par la modulation de leur propre expression et de celle d'autres RBP [
290]. Ils sont également impliqués dans la modulation de la dynamique du SG par interaction avec les ARN cytoplasmiques et autres RBP. La dérégulation des RBP induit également un dysfonctionnement métabolique, le vieillissement et la sénescence [
291 ].
Le transcriptome de la cellule vieillissante entraîne des changements globaux dans l'expression des gènes avec une régulation négative des gènes liés à l'oxydation. respiration, traduction des protéines et signalisation de la croissance, et régulation positive des gènes liés à l'immunité innée, à l'inflammation et aux dommages à l'ADN [
292–
296]. Les multiples couches de traitement qui déterminent l'expression des gènes, y compris la modification de l'ARNm telle que l'épissage, le coiffage et la polyadénylation, l'exportation, la localisation, le renouvellement et la traduction de l'ARN, sont affectées par le vieillissement et la SLA [
297]. Chez plusieurs espèces, y compris les humains et les souris, le vieillissement entraîne des transcriptions d'ARN plus courtes dans près de 80 % des tissus, perturbant l'équilibre des transcriptions d'ARN longues et courtes [
297].
Les voies de signalisation qui contrôlent les alternatives l'épissage font partie des processus les plus dérégulés du vieillissement normal [
292,
298] et la sénescence [
287,
299]. Des taux plus élevés d'épissage alternatif, y compris le rétro-épissage et la formation d'ARN circulaire, une qualité de transcription réduite et des mésappariements avec les séquences du génome sont également détectés au cours du vieillissement [
296]. De plus, au cours du vieillissement naturel, des sites d’épissage cryptiques se révèlent. Il s'agit de séquences dans les introns qui s'incorporent dans la transcription lors de l'épissage, entraînant un codon d'arrêt prématuré et une perte de fonction de la protéine associée [
300].
Le transcriptome vieillissant peut en outre être influencé par une altération de l'ARN. activité polymérase II (Pol II) [
296,
301]. La vitesse d'élongation de l'ARN polymérase II dans les introns augmente avec l'âge dans plusieurs modèles cellulaires et animaux et échantillons humains [
296]. En revanche, le blocage de Pol II au niveau des sites de dommages à l'ADN augmente avec l'âge, ce qui entraîne un stress transcriptionnel et des transcriptions plus courtes [
301]. Les cellules disposent de systèmes rigoureux de contrôle de la qualité de l'ARN pour empêcher ces processus néfastes. Les ARNm épissés de manière aberrante avec des codons d'arrêt prématurés sont dégradés par une « désintégration médiée par le non-sens (NMD) » afin d'empêcher la traduction en protéines délétères non fonctionnelles [
302]. Ce processus est cependant dérégulé avec le vieillissement et affecte particulièrement les neurones post-mitotiques qui sont plus dépendants d'une forte qualité d'ARN. contrôle et processus NMD efficaces [
302].
Les modifications chimiques de l'ARN régulent le métabolisme de l'ARN et sont connues pour contribuer à au moins huit des caractéristiques classiques du vieillissement, notamment la sénescence cellulaire, les changements épigénétiques, les troubles immunitaires et dysfonctionnement des cellules souches, dérégulation métabolique concomitante et perte de protéostase [
303]. Ces modifications d'ARN incluent la méthylation et l'édition A à I. Une diminution des modifications de m6A est présente dans les PBMC humaines âgées [
304], altérant la synthèse des protéines synaptiques et les fonctions synaptiques liées au vieillissement et à la neurodégénérescence [
305], suggérant que la méthylation de l'ARN m6A contribue au déclin cognitif lié au vieillissement [
305]. Édition A-to-I diminue également au cours du vieillissement, spécifiquement dans le cerveau humain [
306] De même, les souris dépourvues de polypeptide catalytique enzymatique éditant l'ARNm de l'apolipoprotéine B (APOBEC1) dans la microglie montrent une accélération de la neurodégénérescence et des déficiences motrices liées à l'âge [
307].
L'expression de divers ARNnc s'altère avec le vieillissement et influence ses cachets. Une gamme d'ARNnc, y compris les ARNnc longs (ARNnc), les microARN (miARN), les ARN interagissant avec les piwis (piARN), les petits ARN nucléolaires (snoARN), les petits ARN nucléaires (snARN), les ARN ribosomiques (ARNr), les petits ARN spécifiques du corps de Cajal. (scaRNA), l'ARN de transfert (ARNt) et les fragments dérivés de l'ARNt (tRF) sont exprimés de manière différentielle dans les tissus vieillissants [
308]. Parmi ceux-ci, les miARN étaient les plus modifiés en raison du vieillissement [
308]. Les changements liés à l'âge dans l'expression globale des gènes sont également en corrélation avec l'expression correspondante des miARN [
309], ce qui n’est pas surprenant étant donné que les miARN régulent l’expression de l’ARNm. Les LncRNA régulent les histones méthyltransférases et d'autres enzymes modifiant la chromatine et modifient ainsi épigénétiquement l'expression des gènes [
310 ]. Ils peuvent également protéger les cellules de la sénescence, car l'ARN non codant (SAN) associé à la sénescence lncRNA a augmenté dans les cellules souches adipeuses (ASC) âgées[
311] et un autre lncRNA, NEAT1, ont supprimé la sénescence cellulaire dans le carcinome hépatocellulaire [
312]. NEAT1 joue un rôle crucial dans la formation d'un environnement flexible au sein des cellules, augmentant les LLPS et la condensation des RBP et des acides nucléiques [
313]. Les CircRNA, une classe plus récemment décrite composée principalement d'ARNnc, sont de plus en plus reconnus comme de puissants régulateurs de l'expression des gènes via leur interaction avec les miARN [
314], et sont des acteurs émergents du vieillissement [
315] et maladies liées à l'âge [
316], y compris la SLA [
317,
318].
Il existe désormais des preuves solides qu'un métabolisme dysfonctionnel de l'ARN est présent dans la SLA. Il est intéressant de noter que les neurones dégénérés dans la SLA présentent des défauts métaboliques de l'ARN (traitement, modifications et transport de l'ARN) similaires à ceux des neurones vieillissants [
298]. Certaines des principales protéines dérégulées dans la SLA sont les RBP, notamment TDP43, FUS, TAF15 et hnRNPA1 [
28,
291]. TDP43 et FUS se localisent mal et s'agrègent dans le cytoplasme dans la SLA sporadique [
319 ], ce qui réduit leur expression dans le noyau [
288], entraînant une perte de fonctions essentielles, notamment l'épissage et la régulation de la transcription. La perte de TDP43 nucléaire conduit également à l’émergence de sites d’épissage cryptiques, qui sont désormais de plus en plus reconnus comme contribuant à l’ALS. Cela inclut un site d'épissage cryptique dans le premier intron du gène stathmin-2303">
303]. Ces modifications d'ARN incluent la méthylation et l'édition A à I. Une diminution des modifications de m6A est présente dans les PBMC humaines âgées [
304], altérant la synthèse des protéines synaptiques et les fonctions synaptiques liées au vieillissement et à la neurodégénérescence [
305], suggérant que la méthylation de l'ARN m6A contribue au déclin cognitif lié au vieillissement [
305]. Édition A-to-I diminue également au cours du vieillissement, spécifiquement dans le cerveau humain [
306] De même, les souris dépourvues de polypeptide catalytique enzymatique éditant l'ARNm de l'apolipoprotéine B (APOBEC1) dans la microglie montrent une accélération de la neurodégénérescence et des déficiences motrices liées à l'âge [
307].
L'expression de divers ARNnc s'altère avec le vieillissement et influence ses cachets. Une gamme d'ARNnc, y compris les ARNnc longs (ARNnc), les microARN (miARN), les ARN interagissant avec les piwis (piARN), les petits ARN nucléolaires (snoARN), les petits ARN nucléaires (snARN), les ARN ribosomiques (ARNr), les petits ARN spécifiques du corps de Cajal. (scaRNA), l'ARN de transfert (ARNt) et les fragments dérivés de l'ARNt (tRF) sont exprimés de manière différentielle dans les tissus vieillissants [
308]. Parmi ceux-ci, les miARN étaient les plus modifiés en raison du vieillissement [
308]. Les changements liés à l'âge dans l'expression globale des gènes sont également en corrélation avec l'expression correspondante des miARN [
309], ce qui n’est pas surprenant étant donné que les miARN régulent l’expression de l’ARNm. Les LncRNA régulent les histones méthyltransférases et d'autres enzymes modifiant la chromatine et modifient ainsi épigénétiquement l'expression des gènes [
310 ]. Ils peuvent également protéger les cellules de la sénescence, car l'ARN non codant (SAN) associé à la sénescence lncRNA a augmenté dans les cellules souches adipeuses (ASC) âgées[
311] et un autre lncRNA, NEAT1, ont supprimé la sénescence cellulaire dans le carcinome hépatocellulaire [
312]. NEAT1 joue un rôle crucial dans la formation d'un environnement flexible au sein des cellules, augmentant les LLPS et la condensation des RBP et des acides nucléiques [
313]. Les CircRNA, une classe plus récemment décrite composée principalement d'ARNnc, sont de plus en plus reconnus comme de puissants régulateurs de l'expression des gènes via leur interaction avec les miARN [
314], et sont des acteurs émergents du vieillissement [
315] et maladies liées à l'âge [
316], y compris la SLA [
317,
318].
Il existe désormais des preuves solides qu'un métabolisme dysfonctionnel de l'ARN est présent dans la SLA. Il est intéressant de noter que les neurones dégénérés dans la SLA présentent des défauts métaboliques de l'ARN (traitement, modifications et transport de l'ARN) similaires à ceux des neurones vieillissants [
298]. Certaines des principales protéines dérégulées dans la SLA sont les RBP, notamment TDP43, FUS, TAF15 et hnRNPA1 [
28,
291]. TDP43 et FUS se localisent mal et s'agrègent dans le cytoplasme dans la SLA sporadique [
319 ], ce qui réduit leur expression dans le noyau [
288], entraînant une perte de fonctions essentielles, notamment l'épissage et la régulation de la transcription. La perte de TDP43 nucléaire conduit également à l’émergence de sites d’épissage cryptiques, qui sont désormais de plus en plus reconnus comme contribuant à l’ALS. Cela inclut un site d'épissage cryptique dans le premier intron du gène stathmin-2 (STMN2), entraînant une perte de protéine et une incapacité à réparer les axones suite à une lésion des motoneurones [
320].
Le contrôle de l'ARN tel que la méthylation de m6A, l'édition A vers I, le NMD et la surveillance de l'ARN sont également dérégulés dans la SLA [
321–
323]. L'expansion de C9ORF72 répète les facteurs d'exportation d'ARN séquestrant [
324 ,
325] et inhibe le NMD [
326]. En revanche, une hyperactivation NMD a été détectée dans les fibroblastes dérivés de patients FUS associés à la SLA [
327. ], qui réduit la biosynthèse des protéines et contribue à la mort des motoneurones dans la SLA [
328]. TDP-43 interagit avec NEAT1, entraînant sa condensation dans des corps nucléaires en réponse au stress et aux caractéristiques pathologiques de la SLA, telles que la phosphorylation et sa mauvaise localisation [
329].
Ces études fournissent donc des preuves significatives reliant le métabolisme dérégulé de l’ARN au vieillissement et à la SLA. Cependant, il est important de reconnaître que ces événements sont étroitement liés [
330 ]. Par exemple, la perte du RBP HuR entraîne une réduction de la méthylation (C106) du composant lncRNA de la télomérase (TERC), ce qui altère la fonction de la télomérase et entraîne une usure des télomères et un vieillissement accéléré [
331] (Tableau
Table 1
Hallmarks of ageing in ALS
| Hallmarks of ageing | Cellular pathways implicated in ALS | Pathological protein involved | References |
|---|
| Primary hallmarks |
| Genomic Instability | Nuclear architecture alterations, nuclear pore pathology, damage to nuclear DNA, damage to mitochondrial DNA | SOD1, TDP-43, C9ORF72 DPRs, FUS, NEK1 | [47, 48, 58, 60, 63, 64, 332, 333] |
| Telomere attrition | Both shorter and longer telomeres are described in ALS | SOD1, C9ORF72 DPRs | [77, 334, 335] |
| Epigenetic alterations | DNA hyper- and hypo-methylation, hyper- or hypo-acetylation of histones | SOD1, FUS, TDP-43, C9ORF72 DPRs | [88, 95–97, 100, 228, 336] |
| Loss of proteostasis | Defects in protein folding, disrupted UPS, defective ER-Golgi trafficking, ER stress, Golgi fragmentation and defects in ERAD | SOD1, TDP-43, C9ORF72 DPRs, FUS | [102, 103, 337–340] |
| Dysregulated macroautophagy | Increased or decreased activation of autophagy, impaired mitophagy, dysregulated autophagy initiation and impaired autophagic flux | C9ORF72 DPRs, SOD1, TDP-43, TBK1, FUS, FIG4, OPTN, UBLN2, SQSTM1, CHMP2B, ALS2 | [341–348] |
| Antagonistic hallmarks |
| Cellular senescence | Microglia senescence, abnormal expression of senescence markers | SOD1, C9ORF72 | [174, 285, 349] |
| Mitochondrial dysfunction | Defects in membrane potential, decreased respiratory capacity, increased free radical production, reduced turnover, and dynamics | SOD1, TDP-43, C9ORF72 DPRs, UBQLN2 and FUS | [74, 191, 196, 332, 350–354] |
| Dysregulated nutrient sensing | Dysregulated mTOR signaling, AMPK pathway and SIRT regulation | SOD1, C9ORF72 DPRs, TDP-43 | [210, 220, 355, 356] |
| Integrative hallmarks |
| Impaired intercellular communication | Dysregulated interaction between glia and neurons, dysregulated EV and intracellular communication | SOD1, TDP-43, FUS and C9ORF72 DPRs | [244, 253, 357, 358] |
| Neuroinflammation | Hyperactivated astrocytes and microglia, increased pro-inflammatory cytokines Changes in glia to proinflammatory phenotypes | SOD1, TDP-43 | [359–361] |
| Stem cell exhaustion | Decreased functionality and regenerative capacity of NSCs | SOD1 | [362] |
| Dysbiosis | Increased intestinal permeability to toxins, altered gut microbiota | SOD1, C9ORF72 DPRs | [283, 284, 286] |
| Defects in RNA dysfunction | RNA metabolic defects (RNA processing, modifications and transport), splicing defects, cryptic exon inclusion, dysregulated quality control of RNA such as NMD and RNA surveillance | TDP43, FUS, TAF15 and hnRNPA1 | [62, 317, 363–366] |
Les caractéristiques décrites ci-dessus détaillent les événements liés au vieillissement au moment niveau léculaire et cellulaire. Ci-dessous, nous discutons également brièvement de la façon dont le vieillissement affecte spécifiquement les types de cellules pertinents pour la SLA ; motoneurones, cellules gliales et cellules musculaires squelettiques.
Il n'est pas clair si les motoneurones sont perdus au cours du vieillissement normal, car des résultats contradictoires ont été obtenus. Certaines études ont conclu que la taille et le nombre de motoneurones restent constants [367], tandis que d'autres rapportent qu'il existe une perte progressive des motoneurones au cours du vieillissement physiologique [105, 368 ], similaire à la SLA, laissant les motoneurones âgés restants sous stress [369].
Des modifications dans la transmission synaptique et l'excitabilité des motoneurones sont l'un des premiers événements de la SLA. L'hyperexcitabilité des motoneurones supérieurs et inférieurs est fréquemment observée dans les modèles de souris SOD1G93A, les motoneurones dérivés de l'ALS-iPSC et les patients SLA [373–375]. L'excitotoxicité, faisant référence à une activation excessive des récepteurs du glutamate et à des lésions neuronales ou à la mort neuronale qui en résulte, est également couramment décrite dans les modèles de maladies [376]. Cependant, au cours de la progression de la maladie, les motoneurones deviennent hypoexcitables, bien que cela puisse être un processus compensatoire [ 376]. Chez les patients SLA, des études électrophysiologiques ont identifié des anomalies des courants sodiques et potassiques, ce qui implique qu'une dépolarisation membranaire et des modifications de l'excitabilité membranaire liées à l'âge sont présentes dans les axones moteurs médians [377]. L'expression accrue de Drosophile dMMP1 dans les motoneurones contribue au déclin de la fonction motrice observé au cours du vieillissement [372]. La surexpression de TDP-43 dans les neurones accélère la mort neuronale en déclenchant l'expression de dMMP1, suggérant des liens potentiels entre le vieillissement et la SLA [372]. Avec le vieillissement, les motoneurones peuvent devenir moins efficaces dans la transmission des signaux aux muscles, ce qui entraîne des temps de réponse plus lents et une diminution du contrôle moteur [378].
Une susceptibilité différentielle entre les sous-types de motoneurones, même au sein d'une même unité motrice, est également observée dans la SLA. Les motoneurones à fatigue rapide (FF) dégénèrent au début de l'évolution de la maladie, et les types résistants à la fatigue (FR) suivent ensuite plus tard. En revanche, les MN lents (S) résistent à la dégénérescence et sont conservés, même jusqu'à un stade avancé de l'évolution de la maladie. Les raisons de la vulnérabilité sélective des sous-types de motoneurones restent floues. Cependant, les sous-types FF et FR sont affectés en premier au cours du vieillissement normal, tandis que les sous-types lents le sont plus tard, ce qui implique que le vieillissement augmente la susceptibilité des motoneurones FF et FR dans la SLA [392–394]. Les sous-types de motoneurones présentent également des différences notables dans leurs propriétés. Les motoneurones FF possèdent des somas de grand diamètre tandis que les motoneurones S contiennent des soma beaucoup plus petits [ 385]. De plus, les motoneurones FF sont beaucoup moins excitable que le sous-type FR qui à son tour est moins excitable que le sous-type S, reliant également la susceptibilité des motoneurones à l'excitabilité. Différents profils d'expression génique sont également évidents entre ces sous-groupes [385].
La combinaison unique de caractéristiques des motoneurones, notamment leurs longs axones, leurs besoins énergétiques élevés, l'augmentation des dommages à l'ADN et la réparation de l'ADN sujette aux erreurs. Les mécanismes neuronaux, la sénescence neuronale, la susceptibilité à l'excitotoxicité et l'hétérogénéité de la susceptibilité entre les sous-types peuvent donc collectivement les rendre plus vulnérables aux effets du vieillissement [395]. Une exploration plus approfondie de ces aspects est cruciale pour comprendre les mécanismes sous-jacents à la dégénérescence des motoneurones liée à l'âge. La caractérisation de ces événements peut ouvrir la voie à des interventions ciblées visant à promouvoir la santé des motoneurones au cours du processus normal de vieillissement.
Les changements liés à l'âge et la présence des signes du vieillissement sont également détecté dans les cellules gliales, comme détaillé ci-dessous. Le rôle de la neuroinflammation induite par les astrocytes réactifs et les microglies est discuté dans la section "Neuroinflammation and aging' de ce document.
Le vieillissement est associé à des changements morphologiques des astrocytes, caractérisés par une atrophie et un rétrécissement de processus tels que les branches et les folioles, et un déclin de leur fonction [396]. Plus précisément, au cours du vieillissement, on observe une diminution des connexions synaptiques et de la plasticité [397]. La protéine acide fibrillaire gliale (GFAP), marqueur des astrocytes, est fortement augmentée dans le cerveau âgé, ce qui représente l'activation et la gliose au cours de la neurodégénérescence [398]. La barrière hémato-encéphalique est également maintenue par les astrocytes, qui deviennent également compromis et fuient au cours du vieillissement [ 398]. La dérégulation de la fonction des astrocytes, conduisant à la libération prolongée de molécules pro-inflammatoires telles que l'IL-8, l'IL-1β, l'IL-6, l'IL-18, le TNF-α, est impliquée dans le vieillissement et les maladies neurogénératives liées à l'âge [399].De nombreuses études ont impliqué les astrocytes dans la pathogenèse de la SLA. Une méta-analyse d'études impliquant des astrocytes humains dérivés d'iPSC avec des variations de SOD1, C9ORF72 et FUS, avec celles utilisant des modèles d'astrocytes de souris exprimant le SOD1G93A une mutation ou une délétion de TDP-43, ou une délétion de Tmem259 (membrane), a révélé un modèle cohérent de changements d'expression génique parmi ces modèles [400], impliquant une régulation positive des gènes associés à la dynamique de la matrice extracellulaire, aux réponses au stress du RE et au système immunitaire [400]. Les astrocytes réactifs induisent l'agrégation des neurofilaments et de la SOD1 en perturbant l'autophagie via l'action du TGF-β1, conduisant à une dégénérescence des motoneurones [401]. Cela se produit par la perturbation de l'autophagie, principalement médiée par le TGF-β1 [401]. Une augmentation du stress oxydatif, une survie réduite des motoneurones et une neurotoxicité ont été observées dans les astrocytes humains dérivés d'iPSC provenant de patients atteints de SLA C9ORF72 [182]. De même, les astrocytes dérivés d'iPSC provenant de patients atteints de SLA FUS R521H, P525L altèrent la croissance des motoneurones-neurites, ainsi que la formation et la fonctionnalité de la formation NMJ [402].
Il existe un débat quant à savoir si l'activation microgliale est présente chez les patients SLA - transition d'une forme ramifiée ou étoilée (état inactif) à une forme forme amiboïde (état actif) – et s'il existe une prolifération accrue et/ou une régulation positive des voies inflammatoires dans les tissus SLA post-mortem [279, 405]. Les preuves provenant de modèles de souris, notamment les knock-outs SOD1G93A, A4V, TDP-43WT, M337V, A315T, FUSWT et C9ORF72, ont révélé une activation des microglies et des changements morphologiques par rapport aux témoins [279, 406– 408]. Les microglies réactives sont neuroprotectrices chez un modèle de souris TDP-43 [409 ]. Les cellules de type microglie dérivées de monocytes présentent les caractéristiques pathologiques de la SLA, notamment l'agrégation cytoplasmique et la phosphorylation du TDP-43, les dommages à l'ADN et l'altération spécifique des cellules de la phagocytose associée à la progression de la maladie [410].
Les cellules de Schwann sont des cellules périphériques génératrices de myéline qui ont une capacité remarquable à se régénérer et à se remyéliniser, mais avec le vieillissement, ces capacités diminuent [417, 418]. Le processus de vieillissement des cellules de Schwann est également lié à des anomalies de la myélinisation chez la souris [418]. De plus, les souris plus âgées présentent une diminution significative du nombre de fibres nerveuses myélinisées [419].
Le vieillissement physiologique peut affecter de manière significative les muscles squelettiques [423], à la fois structurellement et fonctionnellement. Cela peut entraîner une perte progressive de la masse musculaire ou « sarcopénie », entraînant une faiblesse musculaire et un contrôle moteur [423, 424]. La jonction neuromusculaire (NMJ) est une synapse chimique tripartite spécialisée qui implique une communication hautement coordonnée entre le motoneurone présynaptique, le muscle squelettique postsynaptique et les cellules terminales de Schwann. La dénervation musculaire est un contributeur majeur à la sarcopénie [425 ] et les altérations du NMJ au cours du vieillissement jouent un rôle central dans ce processus [425–427] . Dans les muscles, déclin de la fonction mitochondriale lié à l'âge [428] de 25 à 30 % entre 30 et 70 ans [ 429]. De même, des taux de consommation d'oxygène réduits et une production élevée de ROS dans les muscles sont présents au cours du vieillissement normal chez la souris [430].
Alors que le vieillissement normal est une conséquence inévitable pour tous les individus, malgré les Avec l’augmentation de la population âgée, peu d’approches thérapeutiques ont été développées pour le vieillissement lui-même. En revanche, des efforts intenses ont été déployés pour développer des traitements contre les maladies liées à l'âge, notamment la SLA [442]. Des progrès considérables ont été réalisés dans ce domaine et il existe désormais sept médicaments approuvés par la FDA pour la SLA. Cependant, ces traitements ont une efficacité limitée ou sont réservés à des groupes spécifiques de patients. Il est donc important de continuer à développer de nouvelles approches.
Les DNMT, HDAC et HAT ont également été examinés lors du vieillissement. Les HDAC comprennent les désacétylases « classiques » dépendantes de Zn2+ (classes I, II et IV) et les désacétylases SIRT (classe III) qui sont également impliquées dans la longévité [473]. Désactivation des homologues HDAC dans C. elegans et Drosophile une durée de vie améliorée et un déclin physique lié à l'âge retardé [336]. Les inhibiteurs d'HDAC, trichostatine A, scriptaid, tubastatine A, acide valproïque, entinostat, phénylbutyrate de sodium, ont amélioré la durée de vie, retardé l'apparition de la maladie et amélioré la fonction motrice du SOD1G93A souris [101, 472]. Les inhibiteurs d'HDAC pourraient donc avoir un potentiel dans le traitement de maladies liées à l'âge telles que la SLA [474]. Cependant, alors que les inhibiteurs de HDAC le phénylbutyrate de sodium et l'acide valproïque sont sûrs et tolérables dans les essais cliniques de phase II sur la SLA humaine, il n'y a eu aucune différence de survie par rapport au placebo [101].Les stratégies thérapeutiques impliquant la prévention de l'agrégation des protéines et la restauration de la protéostase ont été largement étudiées dans la SLA. Chaperons moléculaires tels que les HSP [475] et PDI les membres de la famille sont neuroprotecteurs contre les pathologies induites par le mutant SOD1 et le TDP-43 de la SLA, y compris in vivo [115, 338, 476] . Les composés ciblant la neuroinflammation se sont révélés largement inefficaces dans les essais cliniques sur la SLA, bien qu'une étude de phase II impliquant le masitinib, un inhibiteur de la tyrosine kinase, associé au riluzole, ait montré des résultats prometteurs chez les patients atteints de SLA [477].
Le phénomène universel du vieillissement est le plus grand facteur de risque de maladies neurodégénératives liées à l'âge, y compris la SLA [335 , 478]. Par conséquent, les stratégies thérapeutiques ciblant les mécanismes du vieillissement peuvent être bénéfiques dans ces conditions. Cependant, à ce jour, le vieillissement n’a pas été exploré spécifiquement en tant que cible de développement de médicaments pour la SLA. De plus, les liens moléculaires entre vieillissement et neurodégénérescence restent mal compris. Le vieillissement normal est un phénomène complexe à plusieurs niveaux, difficile à distinguer de nombreuses maladies associées à l’âge. De plus, les caractéristiques moléculaires et cellulaires reconnues du vieillissement se chevauchent de manière significative et sont fortement liées les unes aux autres (Fig. 1). De plus, il existe de nombreuses similitudes entre le vieillissement et la physiopathologie de la SLA. Cependant, même si la plupart des mécanismes neurodégénératifs impliqués dans la SLA sont liés aux principales caractéristiques du vieillissement, des études supplémentaires sont nécessaires pour confirmer un lien direct entre le dysfonctionnement de ces événements et le vieillissement dans la SLA. La physiopathologie sous-jacente à la SLA implique une interaction complexe entre le vieillissement, des facteurs génétiques, épigénétiques et environnementaux. Comme on pense que la pathogenèse de la SLA est un processus pouvant nécessiter jusqu'à six étapes [479], le vieillissement pourrait donc être un accélérateur important de la neurodégénérescence dans la SLA.
 Ribosomopathies encompass a wide range of syndromes. Common symptoms include a reduced number of blood cells, predisposition to cancer, skeletal abnormalities, and failure to thrive. Ribosomopathy has been associated with skeletal muscle atrophy, which brings us closer to ALS.
Ribosomopathies encompass a wide range of syndromes. Common symptoms include a reduced number of blood cells, predisposition to cancer, skeletal abnormalities, and failure to thrive. Ribosomopathy has been associated with skeletal muscle atrophy, which brings us closer to ALS.
 Alas, the authors resort to an old hypothesis, one of the first about ALS etiology, which is the excitotoxicity hypothesis.
Alas, the authors resort to an old hypothesis, one of the first about ALS etiology, which is the excitotoxicity hypothesis. Cortico-motoneuronal projection is a late evolutionary development that is present only in dexterous primates, such as capuchins, macaques, great apes, and humans, but absent in adult rodents, carnivores, and many others. primates, such as marmosets.
Cortico-motoneuronal projection is a late evolutionary development that is present only in dexterous primates, such as capuchins, macaques, great apes, and humans, but absent in adult rodents, carnivores, and many others. primates, such as marmosets. What is interesting is that 14-3-3θ seems to interact preferentially with pathogenic TDP-43 versions but not with the usual version of TDP-43. This suggests that reducing the production (or increasing the degradation) of 14-3-3θ would reduce the production of pathogenic TDP-43. Scientists have therefore sought to reduce the amount of this 14-3-3θ protein in cells through genetic therapy.
What is interesting is that 14-3-3θ seems to interact preferentially with pathogenic TDP-43 versions but not with the usual version of TDP-43. This suggests that reducing the production (or increasing the degradation) of 14-3-3θ would reduce the production of pathogenic TDP-43. Scientists have therefore sought to reduce the amount of this 14-3-3θ protein in cells through genetic therapy.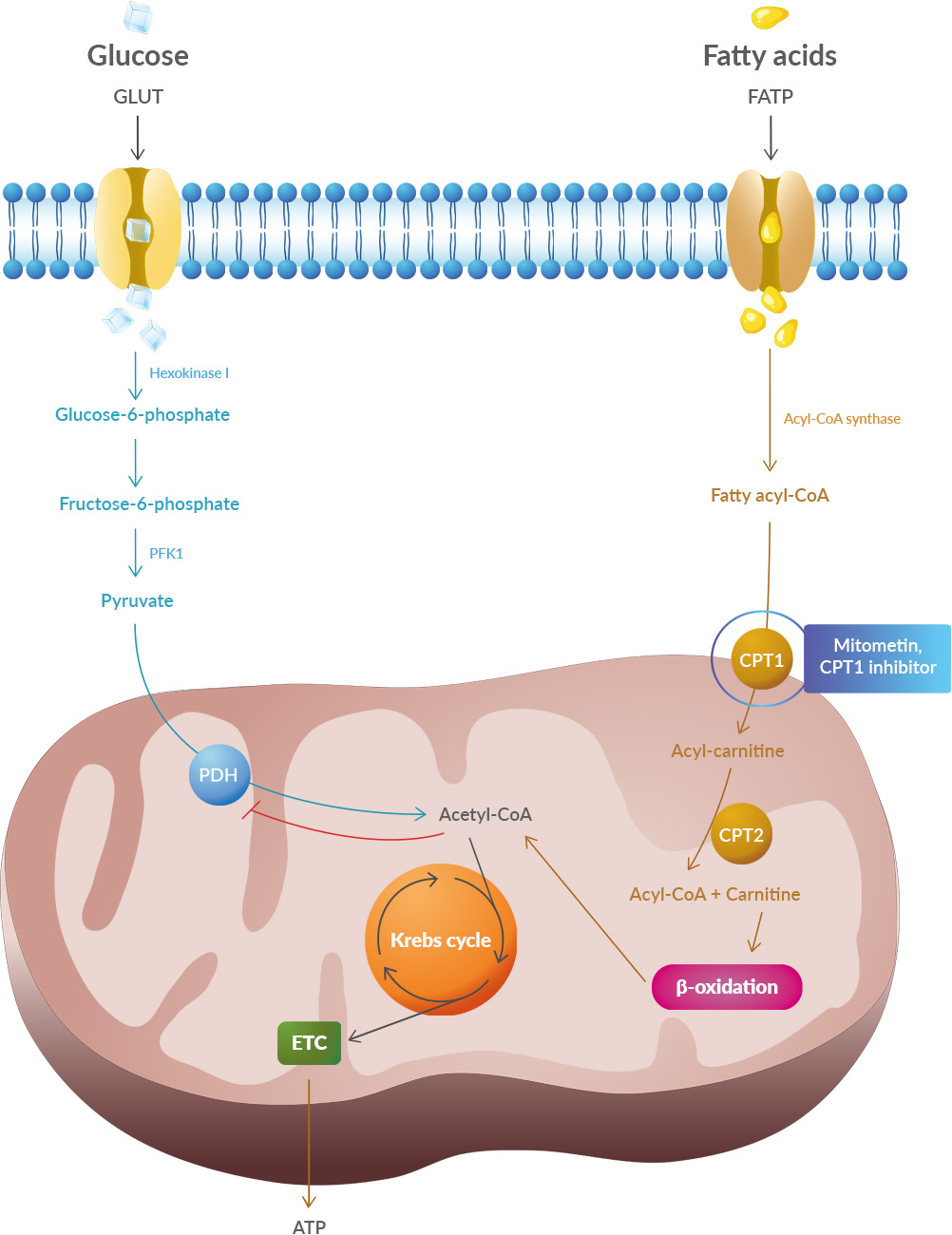 Studies have shown that oxidative stress and endoplasmic reticulum stress are correlated and can lead to protein misfolding (Abramov et al., 2020). Accumulation of misfolded proteins causes cellular damage and mitochondrial dysfunction and is associated with a range of neurodegenerative diseases, including ALS (misfolded SOD1, TDP-43, C9orf72) (McAlary et al., 2020), Parkinson's disease (misfolded α-synuclein) and Alzheimer disease (misfolded Aβ and Tau) (Abramov et al., 2020).
Studies have shown that oxidative stress and endoplasmic reticulum stress are correlated and can lead to protein misfolding (Abramov et al., 2020). Accumulation of misfolded proteins causes cellular damage and mitochondrial dysfunction and is associated with a range of neurodegenerative diseases, including ALS (misfolded SOD1, TDP-43, C9orf72) (McAlary et al., 2020), Parkinson's disease (misfolded α-synuclein) and Alzheimer disease (misfolded Aβ and Tau) (Abramov et al., 2020).
 A new article aims to show that in the case of Zika viruses,
A new article aims to show that in the case of Zika viruses,