Huntington’s Disease and C9orf72 ALS: Shared Mechanisms and Therapeutic Hopes
Approximately 70,000 people have been diagnosed with Huntington’s disease (HD) in the U.S. and Europe, with hundreds of thousands more at risk of inheriting the condition. Despite the clear genetic cause of HD, there are currently no approved therapies that delay onset or slow progression.
Both Huntington's disease and C9orf72-linked ALS, while clinically distinct, share a common hallmark: long, abnormal repetitions of DNA bases. The success of antisense oligonucleotides (ASOs) in spinal muscular atrophy (SMA, SMN1 gene) in 2017, followed by gene therapy in 2019, gave researchers confidence to pursue similar strategies in HD and C9orf72 ALS. Progress in treating one of these repeat expansion diseases may provide hope for others.
1. Genetic Basis
1.1 Huntington’s disease (HD)
HD is caused by an expanded CAG trinucleotide repeat in the HTT gene. - Normal alleles: up to approximately 26 repeats - Pathogenic threshold: 36 or more repeats
CAG encodes glutamine, leading to a mutant protein with an expanded polyglutamine (polyQ) tract. This toxic protein disrupts neuronal function and accumulates throughout the body, contributing not only to neurodegeneration but also to systemic issues like muscle atrophy, cardiac problems, impaired glucose tolerance, weight loss, osteoporosis, and testicular atrophy.

1.2 C9orf72 ALS/FTD
C9orf72-related ALS and frontotemporal dementia (FTD) are caused by an expanded GGGGCC (G4C2) hexanucleotide repeat in the C9orf72 gene. - Normal alleles: up to approximately 30 repeats - Pathogenic alleles: hundreds to thousands
The expansion causes disease through several mechanisms: - Reduced C9orf72 protein levels - Formation of toxic RNA foci - Production of abnormal dipeptide repeat proteins via repeat-associated non-ATG (RAN) translation
1.3 Other repeat expansion diseases
- Spinocerebellar ataxias (SCAs) – many caused by CAG expansions
- Fragile X syndrome – CGG expansion in FMR1
- Myotonic dystrophy – CTG expansion in DMPK
2. Therapeutic Approaches: Shared Strategies
2.1 Antisense oligonucleotides (ASOs)
ASOs aim to reduce toxic transcripts. - HD: ASOs targeting HTT mRNA have reached clinical trials (e.g., Roche/Ionis). - C9orf72 ALS: ASOs targeting repeat-containing transcripts are in early-stage trials.
2.2 Gene silencing/editing
The most advanced approach in HD is uniQure’s AMT-130 gene therapy: - Uses an AAV vector to deliver microRNAs designed to silence mutant HTT. - Administered through MRI-guided stereotactic neurosurgery directly into the striatum. - Clinical trials (U.S. and Europe) are ongoing, with promising early results showing up to 75% slowing in disease progression in high-dose patients over 36 months.
These approaches are not yet cures, but they show that disease modification is possible. Advances in vector design (AAVs, lipid nanoparticles) are directly transferable to other repeat expansion disorders.
2.3 Targeting RNA structures
Small molecules that bind abnormal RNA structures (hairpins, G-quadruplexes) are under development for C9orf72 ALS and myotonic dystrophy, with potential extension to CAG-repeat disorders like HD.
2.4 Modulating protein homeostasis
Strategies to boost autophagy, proteasome activity, or molecular chaperones could reduce toxic protein aggregates in both HD and C9orf72 ALS.
3. Translating Progress Across Diseases
Research tools—such as assays for RNA foci, protein aggregation, and repeat instability—are shared across laboratories working on different repeat expansion disorders. Breakthroughs in one disease can therefore be rapidly tested in others.
Delivery challenges are also common: therapies must reach neurons in the brain and spinal cord. Advances in intrathecal ASO delivery or viral vector engineering benefit all disorders in this family.
In summary: Huntington’s disease and C9orf72 ALS/FTD are distinct conditions, but they share a unifying principle: DNA repeat expansions that disrupt RNA and protein homeostasis. Therapeutic strategies—including antisense oligonucleotides, RNA-targeting drugs, and gene-editing technologies—are broadly applicable across these diseases. Progress in one field accelerates progress in others, offering shared hope for patients facing these devastating neurodegenerative disorders.
 The researchers specifically focused on molecules that could reduce or reverse stress granule formation, particularly those that act directly on stress granule proteins and may be useful as therapeutic agents. From the initial screening, lipoamide emerged as a novel, potent modulator of stress granules.
Once lipoamide was identified as a hit, the researchers sought to determine its effects in cells regarding specificity, potency, intracellular localization, and its effects on other cellular condensates.
The researchers specifically focused on molecules that could reduce or reverse stress granule formation, particularly those that act directly on stress granule proteins and may be useful as therapeutic agents. From the initial screening, lipoamide emerged as a novel, potent modulator of stress granules.
Once lipoamide was identified as a hit, the researchers sought to determine its effects in cells regarding specificity, potency, intracellular localization, and its effects on other cellular condensates.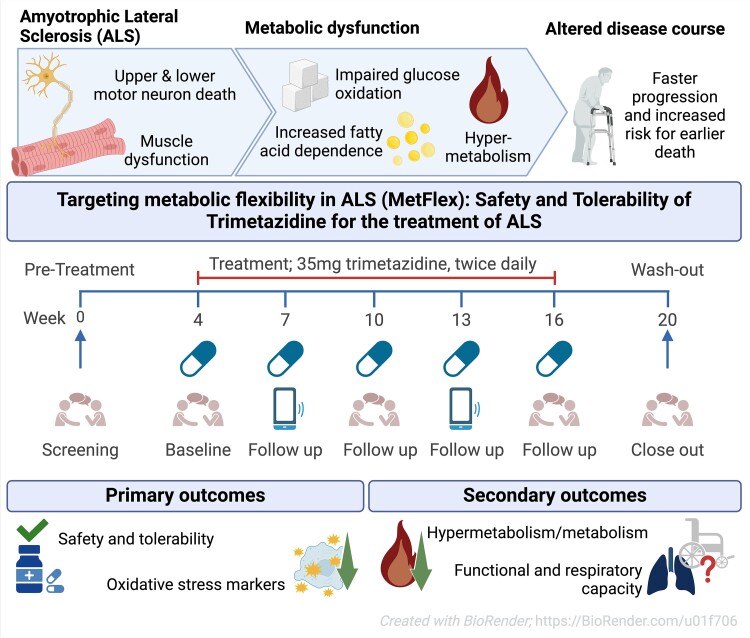 While the publication recounts that trimetazidine was beneficial for patients (this is not a phase III trial), for me the results section does not show conclusive results. For example, the results improved only during the wash-out period.
While the publication recounts that trimetazidine was beneficial for patients (this is not a phase III trial), for me the results section does not show conclusive results. For example, the results improved only during the wash-out period.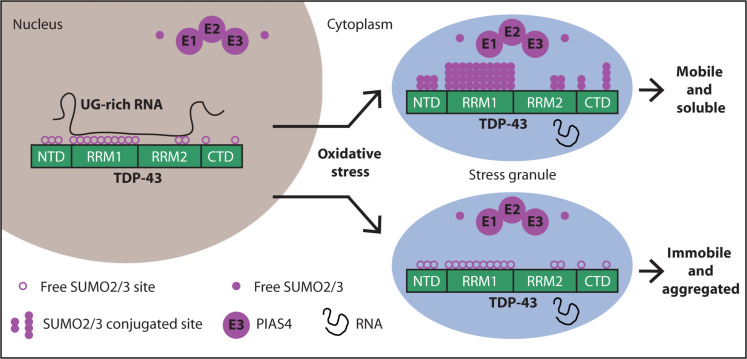 Transactive response DNA-binding protein 43 (TDP-43) is a nuclear RNA binding protein (RBP) involved in RNA metabolism.
TDP-43 has a high propensity to aggregate because of its low solubility in cells and in vitro.
The aggregation propensity of TDP-43 is increased by ALS/FTD-linked mutations and upon exposure to stress and has been observed in patients with C9orf72 hexanucleotide repeat expansion, the most common genetic cause of sporadic and familial.
Transactive response DNA-binding protein 43 (TDP-43) is a nuclear RNA binding protein (RBP) involved in RNA metabolism.
TDP-43 has a high propensity to aggregate because of its low solubility in cells and in vitro.
The aggregation propensity of TDP-43 is increased by ALS/FTD-linked mutations and upon exposure to stress and has been observed in patients with C9orf72 hexanucleotide repeat expansion, the most common genetic cause of sporadic and familial.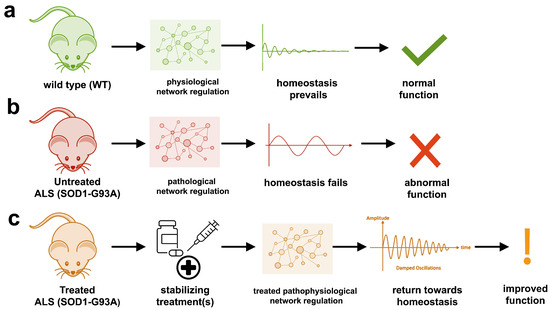 The study uses an innovative and integrative framework to model the regulatory dynamics of wild-type (WT) and SOD1-G93A ALS mice. The models are based on first-order ordinary differential equations (ODEs) that describe how the system output evolves over time. The research uses dynamic meta-analysis to synthesize experimental data from the literature and parameter optimization based on genetic algorithms to infer missing data. Indeed, to build a model, data are needed and here these are obtained from results reported in the literature on SOD1-G93A ALS mouse models.
The study uses an innovative and integrative framework to model the regulatory dynamics of wild-type (WT) and SOD1-G93A ALS mice. The models are based on first-order ordinary differential equations (ODEs) that describe how the system output evolves over time. The research uses dynamic meta-analysis to synthesize experimental data from the literature and parameter optimization based on genetic algorithms to infer missing data. Indeed, to build a model, data are needed and here these are obtained from results reported in the literature on SOD1-G93A ALS mouse models.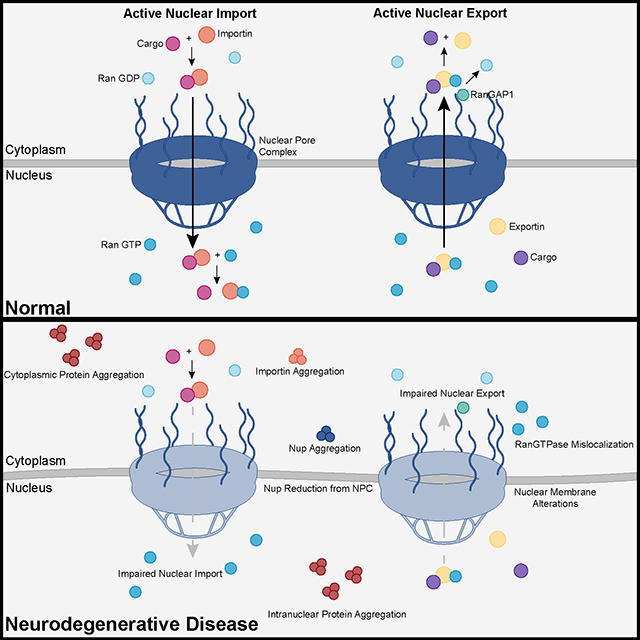 Recent advances into the underlying pathogenic mechanisms have associated mislocalization and aberrant accumulation of disease-related proteins with defective nucleocytoplasmic transport and its mediators called karyopherins.
Recent advances into the underlying pathogenic mechanisms have associated mislocalization and aberrant accumulation of disease-related proteins with defective nucleocytoplasmic transport and its mediators called karyopherins.  They measured somatic repeat expansion over time in individual neurons from donors of different ages. They found that early-phase expansions (e.g., from 40 to 80 CAG repeats) were slow and stochastic, taking decades, while later expansions (e.g., from 80 to 150 repeats) occurred more rapidly.
They then analyzed genetic markers and DNA repair mechanisms associated with repeat instability, such as those involving DNA mismatch repair (MMR) proteins (e.g., MSH3, PMS1). Variants in these genes have been shown to influence the rate of somatic instability. The progression of CAG repeat expansion is driven by errors in DNA replication, repair, and maintenance, particularly in neurons. Key mechanisms include:
They measured somatic repeat expansion over time in individual neurons from donors of different ages. They found that early-phase expansions (e.g., from 40 to 80 CAG repeats) were slow and stochastic, taking decades, while later expansions (e.g., from 80 to 150 repeats) occurred more rapidly.
They then analyzed genetic markers and DNA repair mechanisms associated with repeat instability, such as those involving DNA mismatch repair (MMR) proteins (e.g., MSH3, PMS1). Variants in these genes have been shown to influence the rate of somatic instability. The progression of CAG repeat expansion is driven by errors in DNA replication, repair, and maintenance, particularly in neurons. Key mechanisms include: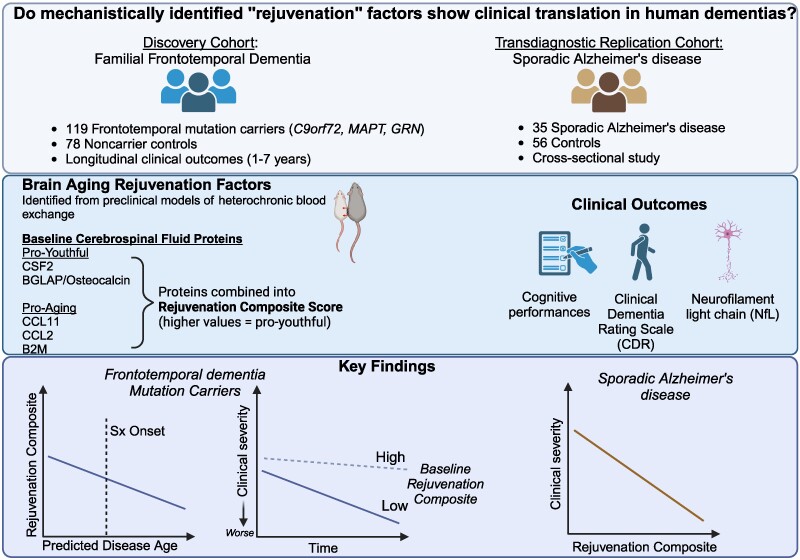 The results appear relatively reliable because the scientists found similar effects in two different types of dementia. The effects were seen across multiple measures (cognitive, functional, and biological markers).
The results appear relatively reliable because the scientists found similar effects in two different types of dementia. The effects were seen across multiple measures (cognitive, functional, and biological markers).