Two recent studies highlight the complex interplay between metabolic dysregulation and ALS progression.
The first study investigated energy balance and glucose control in TAR DNA-binding protein 43 (TDP-43)Q331K mice, which serve as a model for ALS, during both the early and late symptomatic stages of the disease. It suggests the presence of compensatory mechanisms that regulate glucose metabolism differently in this form of ALS. Thus, targeting metabolic pathways, such as insulin signaling and oxidative stress, could provide new therapeutic approaches for ALS.
The etiology of ALS is complex, involving mechanisms such as neuroinflammation, protein aggregation, and energy metabolism dysfunction. The origin of hypermetabolism in amyotrophic lateral sclerosis remains unknown; however, metabolic perturbations in skeletal muscle may be a determining factor, including an increase in the expression of pyruvate dehydrogenase kinase 4 (PDK4), which plays a central role in regulating the oxidation of glucose.
Both sporadic and familial ALS cases commonly exhibit metabolic disturbances, including weight loss, increased resting energy expenditure, and hypermetabolism, which are associated with poorer disease outcomes. Interestingly, a higher body mass index (BMI) at disease onset is linked to increased survival, and high-calorie, high-fat diets have shown some benefits in ALS patients and mouse models, suggesting that metabolic interventions could influence disease progression.
Insulin resistance has been implicated in the progression of ALS. Some studies indicate that diabetes mellitus increases the risk of ALS, while others suggest that type 2 diabetes may delay the onset of the disease. This discrepancy highlights the need for further research into how energy homeostasis and insulin signaling are affected in ALS. Previous studies on SOD1G93A mice, a model of familial ALS, revealed increased energy expenditure and enhanced glucose uptake through insulin-independent pathways, along with glucagon intolerance.
The discrepancy between studies may stem from differences in tissue-specific glucose uptake, as ALS patients exhibit increased glucose uptake in denervated muscles but decreased uptake in the central nervous system.
Building on these findings, the first study investigated metabolic perturbations in the TDP-43Q331K mouse model, which mimics the neuropathological and metabolic hallmarks of human ALS, including TDP-43 pathology, a common feature in both familial and sporadic ALS.
TDP-43Q331K mice exhibited significantly increased daily energy expenditure (DEE) from the early symptomatic stages of the disease. This hypermetabolism was accompanied by a transient increase in food intake, which helped maintain fat mass initially but was insufficient in later stages, leading to fat mass reduction.
During the later stages of the disease, TDP-43Q331K mice showed improved glucose clearance, independent of insulin. Despite reduced circulating glucagon levels, these mice maintained normal fasting blood glucose levels, suggesting alternative mechanisms for glucose regulation.
Unlike SOD1G93A mice, TDP-43Q331K mice did not exhibit insulin or glucagon intolerance. Insulin sensitivity remained unchanged, and while glucagon levels were reduced, the mice maintained normal blood glucose levels, indicating the involvement of other regulatory mechanisms.
Consistent with other ALS models, TDP-43Q331K mice experienced a reduction in lean mass during both early and late disease stages. Regression analysis confirmed that the increased energy expenditure was independent of changes in body mass.
The TDP-43Q331K mutation drives significant metabolic changes, including hypermetabolism and altered glucose uptake, which are not observed with wild-type TDP-43.
The increased glucose uptake in later disease stages is insulin-independent, highlighting the activation of alternative metabolic pathways in response to the disease.
The ability of TDP-43Q331K mice to maintain fasting blood glucose levels despite reduced glucagon suggests the existence of compensatory mechanisms that regulate glucose metabolism differently in this ALS model.
- The second study, a phase 2a clinical trial, explored the pharmacodynamic response of trimetazidine, a partial fatty acid oxidation inhibitor, on oxidative stress markers and energy expenditure in amyotrophic lateral sclerosis. This publication highlights how it's difficult and inconclusive to conduct an ALS clinical trial as twenty-one participants received trimetazidine but only 19 completed the treatment period. While trimetazidine is a well known drug, usually well tolerated, the assessment of energy expenditure may have been uncomfortable. While there were 57 adverse events, the conclusion was, as usual, that the drug was well tolerated!
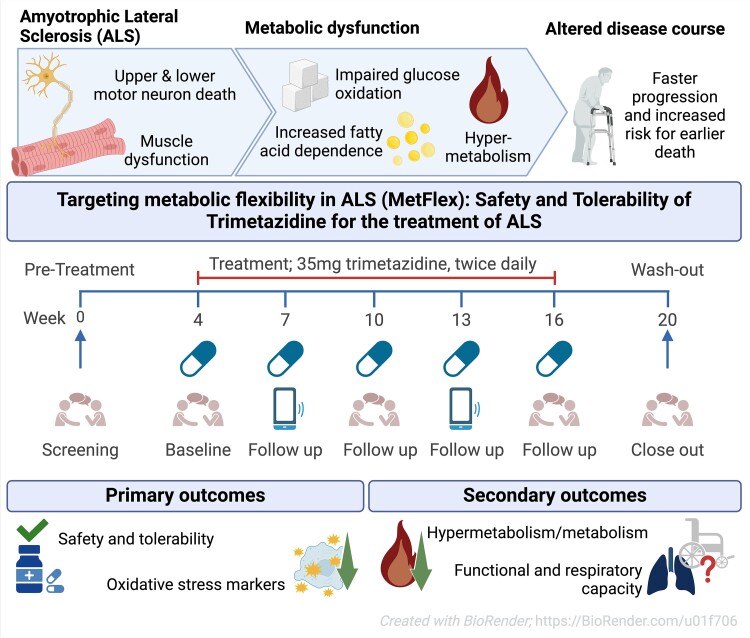 While the publication recounts that trimetazidine was beneficial for patients (this is not a phase III trial), for me the results section does not show conclusive results. For example, the results improved only during the wash-out period.
While the publication recounts that trimetazidine was beneficial for patients (this is not a phase III trial), for me the results section does not show conclusive results. For example, the results improved only during the wash-out period.
The authors tell that the on-treatment period may have been too short, or the sample size too small to detect a disease-relevant change, if one exists. Moreover, in this study, they simply used the approved dose for angina pectoris. Therefore, it remains unclear whether dosing was appropriate and whether a different dose would have resulted in more substantial reductions. Finally, the response in the oxidative stress markers may also be explained by external factors that influence metabolism, such as concomitant medication use and smoking, which cannot be completely ruled out in this uncontrolled study.
While the study was limited by its short duration and lack of a control group, the findings suggest that trimetazidine may help mitigate the hypermetabolic state in ALS and improve disease outcomes. Larger, randomized controlled trials are needed to confirm these results and determine the optimal dosing regimen.
These findings suggest that targeting metabolic pathways, such as insulin signaling and oxidative stress, could offer new therapeutic avenues for ALS. Future research should focus on understanding the underlying mechanisms of metabolic dysregulation, exploring the potential of antidiabetic agents, and conducting larger clinical trials to evaluate the efficacy of metabolic modulators like trimetazidine in ALS patients.