Usually, I strive to find articles that have some interest in patients. This means eliminating the countless articles about cell culture that bring little new knowledge in basic science and have zero interest in pre-clinical research.
Yet here is one about cell culture that may retain some interest. It's about a type of karyopherin that transports protein molecules from the cell's cytoplasm to the nucleus. It does so by binding to specific recognition sequences, called nuclear localization sequences (NLS).
Upon stress, several karyopherin proteins stop shuttling between the nucleus and the cytoplasm and are sequestered in stress granules. Everyone interested in ALS knows this is an important topic in neurodegenerative diseases. Neurodegenerative proteinopathies are characterized by progressive cell loss that is preceded by the mislocalization and aberrant accumulation of proteins prone to aggregation in the cell's cytoplasm.
Despite their different physiological functions, disease-related proteins include tau, α-synuclein, TAR DNA binding protein-43, fused in sarcoma, and mutant huntingtin.
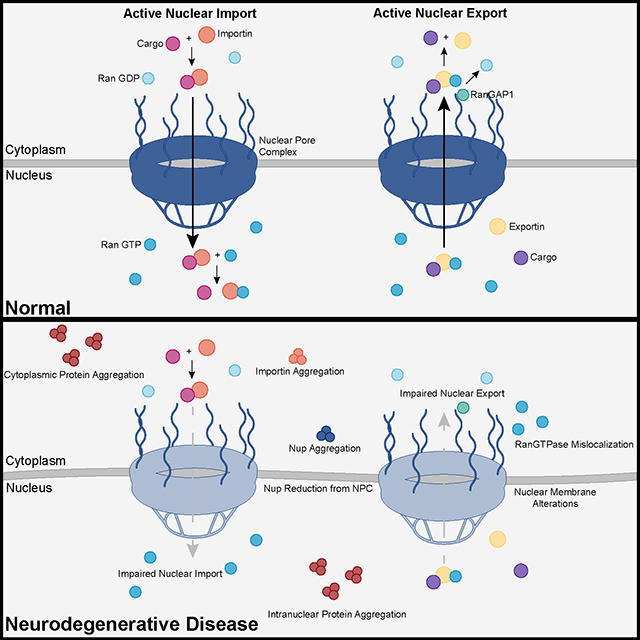 Recent advances into the underlying pathogenic mechanisms have associated mislocalization and aberrant accumulation of disease-related proteins with defective nucleocytoplasmic transport and its mediators called karyopherins. These studies identified karyopherin abnormalities in amyotrophic lateral sclerosis, frontotemporal dementia, Alzheimer’s disease, and synucleinopathies including Parkinson’s disease and dementia with Lewy bodies, that range from altered expression levels to the subcellular mislocalization and aggregation of karyopherin α and β proteins.
Recent advances into the underlying pathogenic mechanisms have associated mislocalization and aberrant accumulation of disease-related proteins with defective nucleocytoplasmic transport and its mediators called karyopherins. These studies identified karyopherin abnormalities in amyotrophic lateral sclerosis, frontotemporal dementia, Alzheimer’s disease, and synucleinopathies including Parkinson’s disease and dementia with Lewy bodies, that range from altered expression levels to the subcellular mislocalization and aggregation of karyopherin α and β proteins.
In addition to their classical function in nuclear import and export, karyopherins can also act as chaperones by shielding aggregation-prone proteins against misfolding, accumulation, and irreversible phase-transition into insoluble aggregates. Karyopherin abnormalities can, therefore, be both the cause and consequence of protein mislocalization and aggregate formation in degenerative proteinopathies.
A new study suggests that the upregulation of two karyopherins might improve cell health in C9orf72.
Hexanucleotide repeat expansions in C9orf72 are the primary genetic mutation associated with amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), collectively referred to as C9-ALS/FTD. The resulting dipeptide repeat (DPR) proteins, such as poly(proline-arginine) (polyPR), generated from G4C2 repeat expansions, have been shown to be highly toxic.
To investigate the cellular localization of PR20, a polyPR protein, it was labeled with fluorescein isothiocyanate (FITC). Notably, several cell lines survived PR20 treatment by sequestering it in the cytoplasm. However, treatment with JQ-1 or Ivermectin (Iver) translocated PR20 into the nucleus, leading to cell death.
Mechanistically, the interaction between KPNA2/KPNB1 and PR20 in the cytoplasm prevented PR20 from entering the cell nucleus. Genetic silencing of KPNA2/KPNB1 converted PR20-resistant cells into PR20-sensitive cells. Specifically, JQ1 treatment decreased KPNA2/KPNB1 protein levels, allowing PR20 to enter the nucleus.
Conversely, overexpressing KPNA2 or KPNB1 effectively blocked cell death induced by co-treatment with JQ-1 and PR20. The authors' findings suggest that the upregulation of KPNA2/KPNB1 protects cells from PR20 toxicity.
Pharmacological targeting of karyopherins represents a promising new strategy for therapeutic intervention in neurodegenerative diseases. Yet by the time patients are diagnosed, a significant number of neurons are already lost, while the remaining functional ones are doomed to degenerate over time due to the lack of efficient treatments. So, it's important not to exaggerate the benefits of drugs in those diseases.
Several compounds targeting karyopherins are already in clinical trials to treat specific forms of cancer and viral infections, so it might be less costly to retarget these drugs toward neurodegenerative diseases than it is for entirely new drugs.