Il y a un lien complexe entre le métabolisme et la myéline lors de la SLA (maladie de Charcot).
Lorsque l'on pense à la SLA (sclérose latérale amyotrophique), les médecins décrivent le plus souvent généralement une maladie neurologique dévastatrice qui affecte les motoneurones, entraînant une faiblesse musculaire et, à terme, une paralysie. Pourtant l'une des meilleurs chances de survie à long terme pour les malades consiste à être en surpoids (IMC: 25).
Des recherches récentes ont révélé des aspects fascinants et moins abordés de la SLA : L'impact sur les muscles arrive en même temps voire avant la dégradation des motoneurones. La peau et d'autres tissus sont également impactés lors de la SLA. Même si peu d'études sont faites à ce sujet, la SLA est caractérisé par un impact profond sur le métabolisme, en particulier celui des lipides (graisses), et ses effets sur la myéline, la gaine protectrice qui entoure les fibres nerveuses. Il est très possible que la dégénérescence des motoneurones et des muscles squeletaux soit due à des anomalies du métabolisme.
La crise énergétique dans la SLA
Chez les personnes en bonne santé, la principale source d'énergie cellulaire est l'ATP, une molécule produite par les cellules principalement via le métabolisme du glucose sanguin. Le glucose est stocké dans le foie et les muscles sous forme de glycogène, puis libéré dans la circulation sanguine par le foie en cas de besoin. Son absorption par les cellules cibles est contrôlée par l'insuline. Cependant, il existe plusieurs voies métaboliques différentes pour générer de l'ATP, outre le métabolisme du glucose.
Des études scientifiques récentes révèlent que de nombreux patients atteints de SLA souffrent d'« hypermétabolisme » :
Leur corps brûle de l'énergie à un rythme nettement supérieur à la normale, même au repos. Environ 50 à 60 % des patients atteints de SLA ont une dépense énergétique au repos 10 à 20 % supérieure à la normale. Ce phénomène est particulièrement surprenant si l'on considère que ces patients perdent progressivement de la masse musculaire, ce qui ralentit généralement leur métabolisme.
Cet hypermétabolisme crée une crise énergétique dans l'organisme. Imaginez que le moteur de votre voiture tourne soudainement à un régime beaucoup plus élevé, même au ralenti : vous consommeriez du carburant beaucoup plus rapidement. De même, les patients atteints de SLA épuisent leurs réserves énergétiques à un rythme accéléré, ce qui contribue à la perte de poids fréquemment observée.
Quand le glucose ne suffit plus : Sources d'énergie alternatives
Que se passe-t-il lorsque votre corps est confronté à une pénurie d'énergie ? Il se met alors à rechercher des sources d'énergie alternatives. Des recherches montrent que dans la SLA, l'organisme se tourne de plus en plus vers les corps cétoniques (dérivés des graisses), qui sont une source d'énergie plus efficace que le glucose (sucre).
Des études ont démontré que les corps cétoniques peuvent générer de l'ATP (la monnaie énergétique des cellules) plus efficacement que le glucose dans certaines conditions. En réponse à la forte demande énergétique, l'organisme des patients atteints de SLA semble mobiliser toutes les sources d'énergie disponibles, notamment les acides gras libres, les triglycérides et les corps cétoniques, pour compenser le déficit énergétique.
Il est intéressant de noter que ces changements métaboliques peuvent survenir avant même l'apparition des symptômes moteurs, ce qui suggère que le dysfonctionnement métabolique pourrait être un signe précoce de la maladie plutôt qu'une simple conséquence.
Lien cholestérol et lésions de la myéline
Au-delà du métabolisme énergétique, la SLA entraîne également d'importantes perturbations du métabolisme du cholestérol. Des études ont révélé des taux de cholestérol élevés chez les patients atteints de SLA par rapport aux personnes en bonne santé. Mais pourquoi est-ce important ?
Le cholestérol est un composant essentiel de la myéline, la gaine protectrice des fibres nerveuses. Dans la SLA, une neurodégénérescence importante entraîne une perte de myéline, particulièrement visible dans le tractus corticospinal, la voie qui transmet les signaux de mouvement du cerveau à la moelle épinière. Ce processus libère du cholestérol, qui doit être stocké ou éliminé du système nerveux central.
Des recherches récentes sur des modèles murins C9orf72 (C9orf72 est la cause génétique la plus fréquente de la SLA) montrent que lorsque ce système d'élimination du cholestérol est défaillant, un environnement toxique se crée qui accélère la progression de la maladie. L'organisme tente de gérer l'excès de cholestérol par plusieurs mécanismes :
L'excès de cholestérol est converti en esters de cholestérol et stocké dans les gouttelettes lipidiques à l'intérieur des cellules, notamment dans les cellules cérébrales appelées oligodendrocytes (cellules productrices de myéline).
Des protéines comme ApoE et Abca1, qui contribuent à l'élimination du cholestérol, sont régulées à la hausse.
L'organisme diminue la production de nouveau cholestérol pour équilibrer l'excès.
Lorsque ces mécanismes échouent ou sont surchargés, le cholestérol et ses dérivés peuvent devenir toxiques pour les cellules nerveuses et les oligodendrocytes qui les soutiennent.
Oligodendrocytes associés à une maladie (OLD)
La principale fonction des Oligodendrocytes est la formation de la gaine de myéline entourant les fibres nerveuses (axones) du système nerveux central.
L'une des découvertes récentes les plus intrigantes est l'identification d'un type spécifique d'oligodendrocyte dysfonctionnel qui apparaît dans la SLA et d'autres maladies neurodégénératives. Ces « oligodendrocytes associés à la maladie » (OLD) présentent un profil d'expression génétique caractéristique qui reflète leur état de stress.
Dans les modèles de SLA, ces OLD semblent contribuer à la progression de la maladie en ne maintenant pas une myéline adéquate et en libérant potentiellement des substances nocives. Une protéine appelée PLIN4, qui enrobe les gouttelettes lipidiques, est fortement augmentée dans ces cellules, servant de marqueur moléculaire de ce dysfonctionnement.
Le lien avec l'inflammation
L'excès de cholestérol n'affecte pas seulement directement les neurones et les oligodendrocytes. Il influence également la réponse immunitaire cérébrale. La microglie, les cellules immunitaires du cerveau, s'active et prend l'apparence de « cellules spumeuses » lorsqu'elle tente d'absorber et d'éliminer les débris de myéline riches en cholestérol.
Ce processus peut déclencher une inflammation par différentes voies :
- Les cristaux de cholestérol peuvent activer un terrain inflammatoire
- Les dérivés auto-oxydés du cholestérol peuvent endommager les motoneurones
- Le processus de clairance lui-même peut produire des sous-produits toxiques
Cette réponse inflammatoire peut endommager davantage les neurones, créant Un cercle vicieux de dégénérescence.
Interventions métaboliques : une nouvelle frontière thérapeutique ?
Ces connaissances sur le métabolisme de la SLA ouvrent de nouvelles perspectives thérapeutiques prometteuses, ciblant à la fois le métabolisme énergétique et la gestion du cholestérol.
Approches axées sur l'énergie
Des essais cliniques explorent actuellement des interventions nutritionnelles. L'étude LIPCAL-ALS a révélé que des compléments alimentaires riches en calories et en graisses présentaient des effets bénéfiques sur la survie et les marqueurs de progression de la maladie chez les patients dont la maladie progresse rapidement. D'autres études étudient la supplémentation en corps cétoniques comme autre approche pour combler le déficit énergétique.
Approches axées sur le cholestérol
Un médicament appelé (CD), qui aide à séquestrer l'excès de cholestérol, montre des résultats prometteurs dans des modèles murins de SLA. Chez les souris femelles porteuses de la mutation C9orf72, le traitement par CD a prolongé leur durée de vie, réduit les marqueurs de neurodégénérescence, amélioré la myélinisation et modifié la réponse microgliale nocive en une réponse plus bénéfique.
Il est intéressant de noter que la cyclodextrine est déjà utilisée comme excipient dans de nombreuses formulations pharmaceutiques et est actuellement testé dans le cadre d'essais cliniques pour d'autres pathologies, comme la maladie de Niemann-Pick de type C et la maladie d'Alzheimer. Il représente une opportunité potentielle de réorientation pour le traitement de la SLA.
Conclusion
Les aspects métaboliques et myéliniques de la SLA révèlent que cette maladie affecte bien plus que les motoneurones : elle perturbe les systèmes énergétiques fondamentaux de l'organisme et l'infrastructure essentielle au bon fonctionnement des nerfs.
En comprenant ces changements métaboliques et l'interaction complexe entre le métabolisme énergétique, la gestion du cholestérol et l'inflammation, les chercheurs espèrent développer de nouvelles thérapies susceptibles de ralentir la progression de la maladie et d'améliorer la qualité de vie des patients.
Bien que ces interventions métaboliques ne guérissent probablement pas la SLA, elles constituent une approche complémentaire importante pour traiter cette maladie complexe. À mesure que la recherche progresse, les liens entre le métabolisme, la santé de la myéline et la neurodégénérescence révéleront probablement encore plus de cibles thérapeutiques potentielles.
 What should we consider about all this? Maybe we should ask why scientists are searching for new drugs instead of focusing on compounds of drugs that have already shown some effects. Perhaps everyone wants to get rich, so they avoid exploring drugs that can't be patented.
What should we consider about all this? Maybe we should ask why scientists are searching for new drugs instead of focusing on compounds of drugs that have already shown some effects. Perhaps everyone wants to get rich, so they avoid exploring drugs that can't be patented. Les progrès réalisés dans la chimie médicinale des ASO ont amélioré leur profil pharmacodynamique, permettant ainsi une administration sûre et efficace au système nerveux central. Les thérapies ASO pour la SLA se sont rapidement développées au cours des deux dernières décennies, et les ASO ciblant SOD1, C9orf72 et ATXN2 sont actuellement en essais cliniques pour les formes familiales ou sporadiques de SLA.
Les progrès réalisés dans la chimie médicinale des ASO ont amélioré leur profil pharmacodynamique, permettant ainsi une administration sûre et efficace au système nerveux central. Les thérapies ASO pour la SLA se sont rapidement développées au cours des deux dernières décennies, et les ASO ciblant SOD1, C9orf72 et ATXN2 sont actuellement en essais cliniques pour les formes familiales ou sporadiques de SLA. Identifié par criblage in vitro, l'ASO ION363 développé par la société IONIS qui a aussi développé Tofersen, cible le 6e intron de FUS (SLA avec une mutation P525L).
ION363 réduit les taux de protéines de liaison à l'ARN insolubles et insolubles associées aux agrégats, telles que hnRNPA1 et ralentit la neurodégénérescence des motoneurones lombaires et la perte d'innervation de la jonction neuromusculaire.
Identifié par criblage in vitro, l'ASO ION363 développé par la société IONIS qui a aussi développé Tofersen, cible le 6e intron de FUS (SLA avec une mutation P525L).
ION363 réduit les taux de protéines de liaison à l'ARN insolubles et insolubles associées aux agrégats, telles que hnRNPA1 et ralentit la neurodégénérescence des motoneurones lombaires et la perte d'innervation de la jonction neuromusculaire. The researchers specifically focused on molecules that could reduce or reverse stress granule formation, particularly those that act directly on stress granule proteins and may be useful as therapeutic agents. From the initial screening, lipoamide emerged as a novel, potent modulator of stress granules.
Once lipoamide was identified as a hit, the researchers sought to determine its effects in cells regarding specificity, potency, intracellular localization, and its effects on other cellular condensates.
The researchers specifically focused on molecules that could reduce or reverse stress granule formation, particularly those that act directly on stress granule proteins and may be useful as therapeutic agents. From the initial screening, lipoamide emerged as a novel, potent modulator of stress granules.
Once lipoamide was identified as a hit, the researchers sought to determine its effects in cells regarding specificity, potency, intracellular localization, and its effects on other cellular condensates.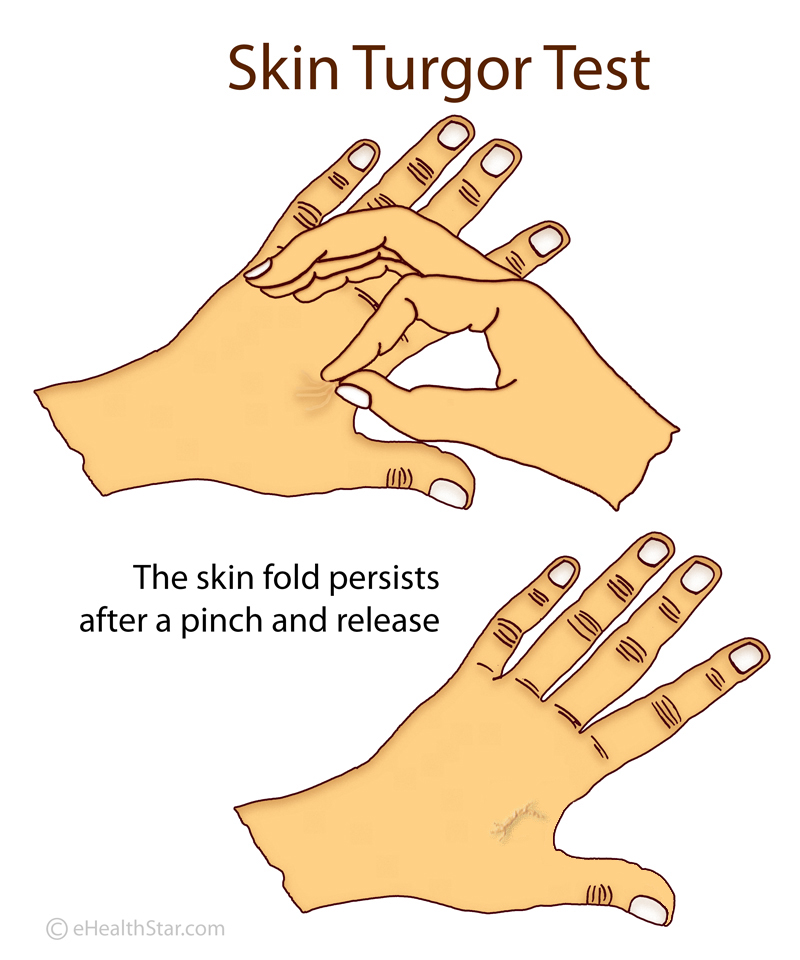 In healthy individuals, after a deformation or pinching, the skin quickly returns to its original shape. In patients with ALS, this return is slower. This is called the delayed return phenomenon (DRP).
In healthy individuals, after a deformation or pinching, the skin quickly returns to its original shape. In patients with ALS, this return is slower. This is called the delayed return phenomenon (DRP). Some people in the intervention group showed an astonishingly slower disease progression compared to the delayed-start group:
Some people in the intervention group showed an astonishingly slower disease progression compared to the delayed-start group: