A few years ago I tried to model the course of ALS using SBML, a systems biology tool now abandoned by scientists.
At the time there was no model of ALS, but since then there have been very interesting attempts, for example, this one:enter link description here
However, few scientists and almost no doctors use models based on differential equations today, we live in an era where statistical models are all the rage. In addition, biology as it is practiced is essentially qualitative, which makes it not very suitable for modeling and which attracts the mockery of "soft science", because by nature it is not capable of making consistent predictions. One only has to look at the colossal failure rate of clinical trials to be convinced of this.
The publication reviewed in this post focuses on understanding the regulatory dynamics of amyotrophic lateral sclerosis (ALS) using the widely used SOD1-G93A transgenic mouse model.
ALS is a multifactorial disease, and previous studies have often focused on isolated aspects rather than its complex and interconnected nature.
The study suggests that ALS regulation may be hypervigilant, meaning that the system overcorrects in response to stress, leading to damaging oscillatory behavior that contributes to disease progression.
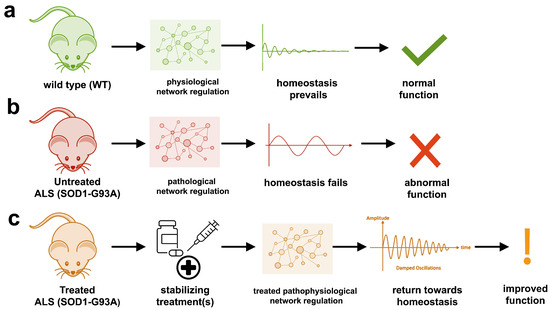 The study uses an innovative and integrative framework to model the regulatory dynamics of wild-type (WT) and SOD1-G93A ALS mice. The models are based on first-order ordinary differential equations (ODEs) that describe how the system output evolves over time. The research uses dynamic meta-analysis to synthesize experimental data from the literature and parameter optimization based on genetic algorithms to infer missing data. Indeed, to build a model, data are needed and here these are obtained from results reported in the literature on SOD1-G93A ALS mouse models.
The study uses an innovative and integrative framework to model the regulatory dynamics of wild-type (WT) and SOD1-G93A ALS mice. The models are based on first-order ordinary differential equations (ODEs) that describe how the system output evolves over time. The research uses dynamic meta-analysis to synthesize experimental data from the literature and parameter optimization based on genetic algorithms to infer missing data. Indeed, to build a model, data are needed and here these are obtained from results reported in the literature on SOD1-G93A ALS mouse models.
The study shows that SOD1-G93A mice with ALS exhibit unstable physiological regulation, characterized by oscillatory behavior due to hypervigilant regulation. This instability intensifies near disease onset and worsens with progression.
Computational models of the physiological dynamics of wild-type (WT) and transgenic SOD1-G93A mice were constructed: a WT mouse model to simulate normal homeostasis and a SOD1-G93A ALS model to simulate the dynamics of ALS pathology and their response to treatments in silico. The model simulates the functional molecular mechanisms of apoptosis, metal chelation, energetics, excitotoxicity, inflammation, oxidative stress, and proteomics using data curated from published SOD1-G93A mouse experiments. Time-course measures of disease progression (rotarod, grip strength, body weight) were used to validate the results from the literature.
The health of untreated SOD1-G93A ALS mouse models cannot be maintained due to oscillatory instability. The onset and magnitude of homeostatic instability corresponded with disease onset and progression. Oscillations are associated with high feedback gain due to hypervigilant regulation.
Multiple virtual treatments combined were able to stabilize the dynamics of SOD1-G93A ALS mice to near-normal WT homeostasis. However, treatment timing and effect size are critical for stabilization to match therapeutic success. The most common unidirectional stabilizing treatment was pro-proteomics, while the most common bidirectional stabilizing treatment was energy consumption and anti-apoptosis. The authors cite anti-apoptosis factors such as caspase-9 inhibitor, caspase-3 inhibitor, Bax inhibitor, and Bcl-2 homolog BCL-XL.
A major drawback is that this type of ALS only affects less than 2% of ALS cases and if it is still largely overrepresented in the literature, it is because the association of this type of mutation with ALS is historically the first to have been discovered during the boom in genetics and for almost 15 years no other association has been found.
Another important point is that we do not know what mechanisms cause this disease, or even if it is a single disease. Models of this type therefore make many assumptions and their results are less robust than they appear.
The study highlights the multifactorial nature of ALS, which involves various molecular mechanisms, such as apoptosis, bioenergetics, excitotoxicity, inflammation, oxidative stress, and proteomics. These mechanisms are interconnected, and their dysregulation contributes to disease progression.
The study also explores the potential of combination therapies to stabilize the regulatory dynamics of ALS. Previous studies focusing on single therapeutic targets have often been inadequate, but combination treatments have shown success in the management of other complex diseases such as cancer, COVID-19, and HIV. The study suggests that precisely timed combination therapies targeting multiple pathways may be necessary to achieve meaningful results in ALS.
Code and data are not available at the indicated Github address.
The authors acknowledge that first-order feedback models may not fully capture the complexity of biological systems, including phenomena such as bistability and hysteresis. But for now, we are still in the prehistory of biological systems modeling, the field needs to be developed before focusing on its imperfections.
I fear that this publication will go way over the heads of health professionals working in the field of ALS.
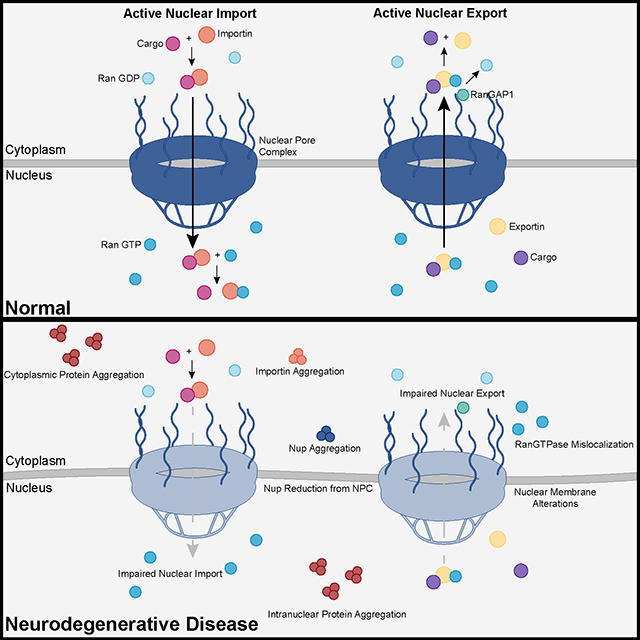 Recent advances into the underlying pathogenic mechanisms have associated mislocalization and aberrant accumulation of disease-related proteins with defective nucleocytoplasmic transport and its mediators called karyopherins.
Recent advances into the underlying pathogenic mechanisms have associated mislocalization and aberrant accumulation of disease-related proteins with defective nucleocytoplasmic transport and its mediators called karyopherins.  A
A  They measured somatic repeat expansion over time in individual neurons from donors of different ages. They found that early-phase expansions (e.g., from 40 to 80 CAG repeats) were slow and stochastic, taking decades, while later expansions (e.g., from 80 to 150 repeats) occurred more rapidly.
They then analyzed genetic markers and DNA repair mechanisms associated with repeat instability, such as those involving DNA mismatch repair (MMR) proteins (e.g., MSH3, PMS1). Variants in these genes have been shown to influence the rate of somatic instability. The progression of CAG repeat expansion is driven by errors in DNA replication, repair, and maintenance, particularly in neurons. Key mechanisms include:
They measured somatic repeat expansion over time in individual neurons from donors of different ages. They found that early-phase expansions (e.g., from 40 to 80 CAG repeats) were slow and stochastic, taking decades, while later expansions (e.g., from 80 to 150 repeats) occurred more rapidly.
They then analyzed genetic markers and DNA repair mechanisms associated with repeat instability, such as those involving DNA mismatch repair (MMR) proteins (e.g., MSH3, PMS1). Variants in these genes have been shown to influence the rate of somatic instability. The progression of CAG repeat expansion is driven by errors in DNA replication, repair, and maintenance, particularly in neurons. Key mechanisms include: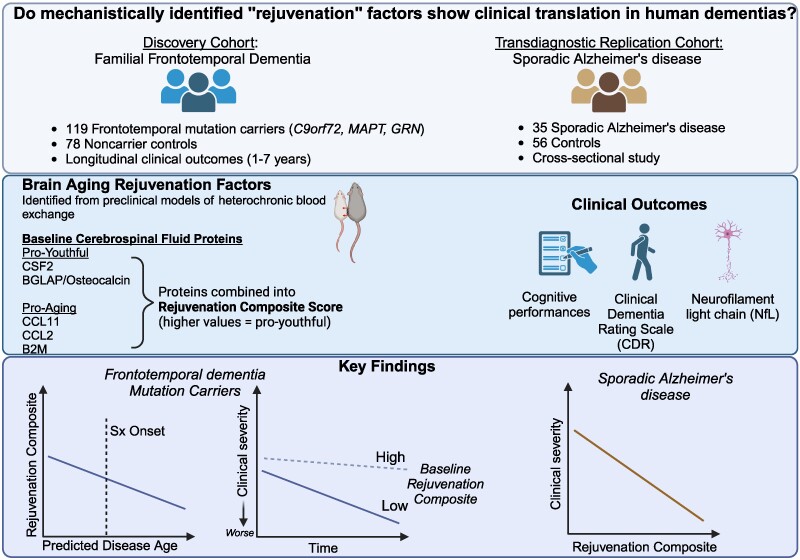 The results appear relatively reliable because the scientists found similar effects in two different types of dementia. The effects were seen across multiple measures (cognitive, functional, and biological markers).
The results appear relatively reliable because the scientists found similar effects in two different types of dementia. The effects were seen across multiple measures (cognitive, functional, and biological markers). The endoplasmic reticulum (ER) is an important organelle in cells that is involved in protein conformation. This step occurs after protein synthesis by ribosomes and after conformation, the new protein will be sent to its final destination by the Golgi apparatus. Protein conformation requires energy, so when disease occurs, the ER may not be able to properly conform the new proteins.
The endoplasmic reticulum (ER) is an important organelle in cells that is involved in protein conformation. This step occurs after protein synthesis by ribosomes and after conformation, the new protein will be sent to its final destination by the Golgi apparatus. Protein conformation requires energy, so when disease occurs, the ER may not be able to properly conform the new proteins. Throughout the study, incidence rates ranged from 2/100,000 person-years in 2008 to 2.77/100,000 in 2021), with the latter year being the year with the highest incidence recorded.
Throughout the study, incidence rates ranged from 2/100,000 person-years in 2008 to 2.77/100,000 in 2021), with the latter year being the year with the highest incidence recorded.