In the case of neurodegenerative diseases, it is certainly more useful to try to develop new neuronal cells than to "cure" cells that we are told are dying or even dead for months. This is the purpose of regenerative medicine.
One of the difficulties is that neurons are cells that do not reproduce, we live with the stock that we had at the end of our puberty. These cells come from stem cells.
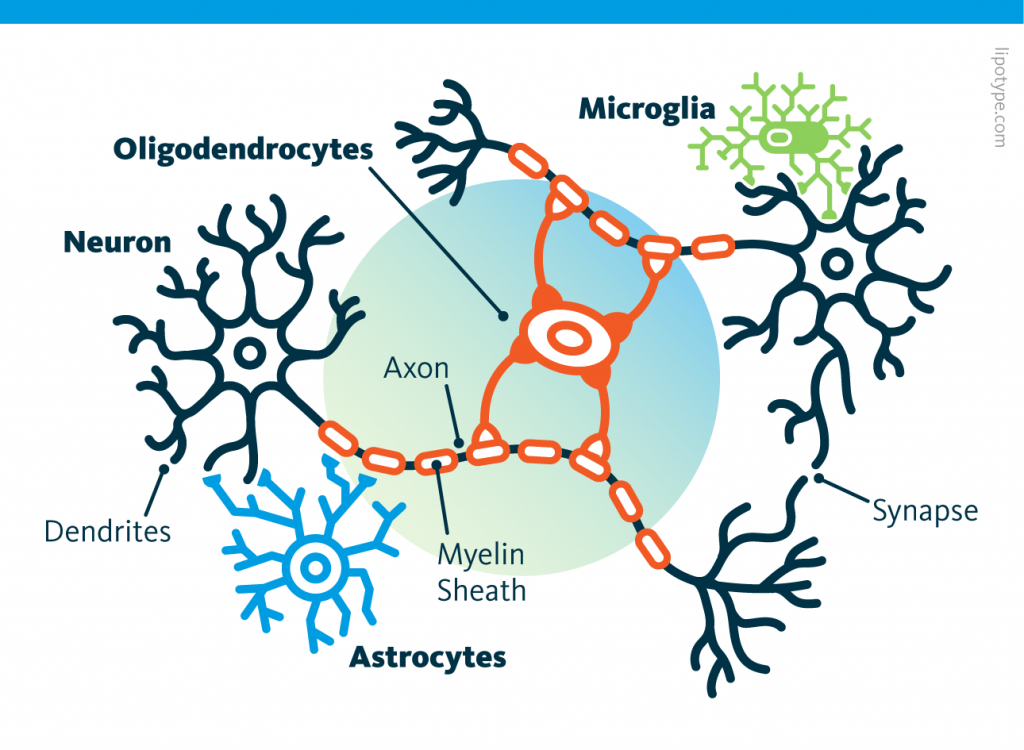
In our brain and the rest of the central nervous system, half of the cells are not neurons, but more classic cells that nourish and maintain neurons.
These cells reproduce by fission like most cells. In particular, astrocytes, although tiny, have many points in common with neurons. It is therefore natural to want to transform astrocytes into neuronal stem cells.
 Curiously, scientists are rather looking to directly convert astrocytes into neurons, despite the enormous morphological difference between these two types of cells.
Curiously, scientists are rather looking to directly convert astrocytes into neurons, despite the enormous morphological difference between these two types of cells.
A few years ago, following the discovery of Yamanaka factors, it was believed that this goal could be achieved quickly. However, the results obtained have been contested, in particular, because of methodological flaws. Despite the promising results of previous studies, the lack of robust lineage tracing methods has led to uncertainty as to whether the induced neurons originated from glial cells or whether native neurons were mistakenly labeled.
A new publication describes recent advances in converting glial cells into induced neurons in the central nervous system, particularly in regions where stem cell activity is limited. The previous “lineage reprogramming” approach used transcription factors to change the identity of terminally differentiated cells, such as glial cells, to other cell types.
The current study focuses on the transcription factor Ascl1, known for its role in neuronal fate decisions and its potential to reprogram various cell types into neurons. While Ascl1 can efficiently convert glial cells into induced neurons in vitro, its efficacy in vivo varies widely, often leading to limited neurogenic effects or, in some cases, inducing glial proliferation rather than neuronal conversion.
This inconsistency suggests that regulatory challenges affect Ascl1’s performance as a reprogramming factor, particularly due to context-specific modifications such as phosphorylation, which modulate its activity. Previous research has found that a mutant variant of Ascl1, known as Ascl1SA6, engineered to resist phosphorylation, enhances neurogenic activity, prompting this study to determine whether Ascl1SA6 could enhance glia-to-neuron reprogramming in vivo.
The researchers tested Ascl1SA6 in the early postnatal mouse cerebral cortex, which showed superior neuronal induction capacity compared to wild-type Ascl1. Furthermore, the combination of Ascl1SA6 with the survival factor Bcl2 further enhanced the efficiency of iN conversion. The study’s lineage tracing confirmed that the newly formed induced neurons were primarily from astrocytes. Interestingly, many of these induced neurons exhibited characteristics of fast-spiking parvalbumin-positive (PV+) interneurons.
In summary, the study demonstrates that Ascl1SA6, particularly in combination with Bcl2, could improve the efficiency and fidelity of glia-to-neuron reprogramming in vivo, with potential implications for therapies targeting neuronal loss and brain circuit restoration.
It should be kept in mind, however, that these cell type conversions a priori promote the appearance of cancers, that the new neurons are hardly useful, that the method used is gene therapy and therefore has very low efficacy and elevates cancer risks, that is is a report on experiences done in mice and they usually do not translate in humans and that it is not a question of conversion to neuronal stem cells which would be free of many of these problems.
Nevertheless, it's a step in the right direction.
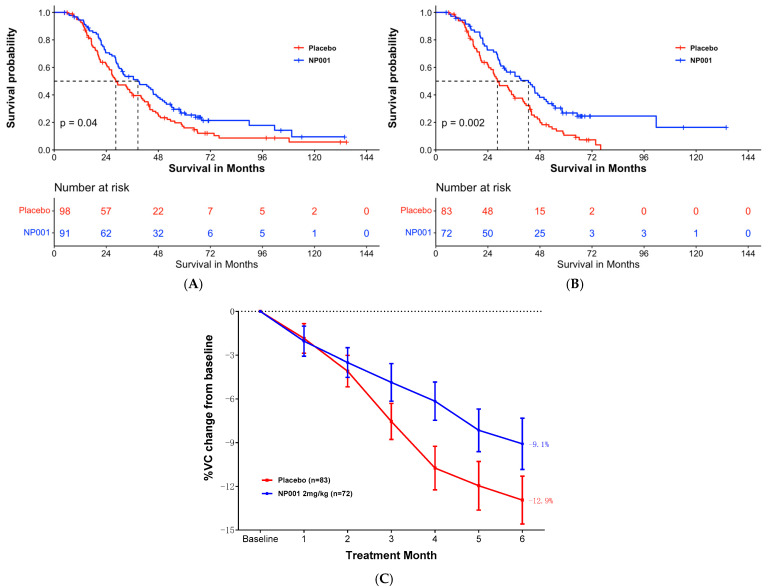 For example, the authors claim that survival of ALS patients with inflammation is 16 months longer than in the placebo arm. At 72 months there were only 3 people in the inflammation subset and two people in the placebo arm. Nobody can say anything about these numbers. If we use the same criteria, it shows that NP001 worsened the condition in all pALS with respect to the placebo branch, as starting from 72 months there were fewer survivors in the NP001 arm than in the placebo.
For example, the authors claim that survival of ALS patients with inflammation is 16 months longer than in the placebo arm. At 72 months there were only 3 people in the inflammation subset and two people in the placebo arm. Nobody can say anything about these numbers. If we use the same criteria, it shows that NP001 worsened the condition in all pALS with respect to the placebo branch, as starting from 72 months there were fewer survivors in the NP001 arm than in the placebo. The authors observed that, despite a decrease in limb motor functions, the feeding function of these mice was preserved until the late stages of the disease. Remarkably, the masseter muscle showed no reduction in muscle volume, wet weight, or muscle fiber cross-sectional area. Furthermore, no changes were observed in muscle fiber types, indicating a possible resistance of the masseter muscle to ALS-induced impairment. A potential reason for the lack of atrophy in the masseter muscle could be its higher number of muscle satellite cells compared to that of the gastrocnemius muscle. This abundance may promote the maintenance of muscle fiber nuclei, thereby contributing to muscle tissue regeneration.
The authors observed that, despite a decrease in limb motor functions, the feeding function of these mice was preserved until the late stages of the disease. Remarkably, the masseter muscle showed no reduction in muscle volume, wet weight, or muscle fiber cross-sectional area. Furthermore, no changes were observed in muscle fiber types, indicating a possible resistance of the masseter muscle to ALS-induced impairment. A potential reason for the lack of atrophy in the masseter muscle could be its higher number of muscle satellite cells compared to that of the gastrocnemius muscle. This abundance may promote the maintenance of muscle fiber nuclei, thereby contributing to muscle tissue regeneration. Those other cells, which compose half of the brain's cells, are receiving more attention. There are multiple types but normally they are there to assist neurons in their task. A simplified view tells that neurons are a sort of plumbing system and the glial cells are the real actors in the brain.
Those other cells, which compose half of the brain's cells, are receiving more attention. There are multiple types but normally they are there to assist neurons in their task. A simplified view tells that neurons are a sort of plumbing system and the glial cells are the real actors in the brain.