One of the most devastating for of amyotrophic lateral sclerosis (ALS) is the loss of control of the muscles involved in speech and swallowing, which is called "bulbar onset". Early detection of changes during or after an ALS diagnosis could pave the way for interventions that improve quality of life, but conventional assessments often rely on subjective observation.
Indeed, surface electromyography observes the reaction of muscles that are excited by an electric current. This observation is subjective, it involves interpreting electrical signals by identifying patterns that are not obvious because they are fleeting and non-standardized. This observation depends heavily on the placement of the needles, but above all, from this observation the practitioner deduces the good or bad functioning of the lower and upper motor neurons. I am not a doctor, just a retired engineer, but I find that these deductions of the functioning of motor neurons from surface electromyography make absolutely no sense, no logic. Yet the majority of ALS diagnoses are made this way!
Furthermore, this approach is poorly adapted to bulbar involvement. Also, a slightly more standardized method of interpreting surface electromyography adapted to bulbar involvement would be welcome.
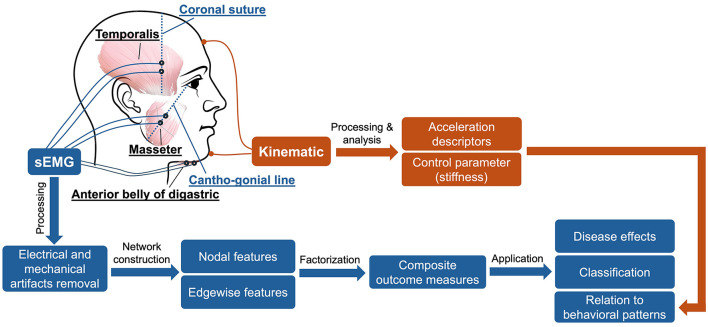 A recent study renovates surface electromyography by automatically recording and interpreting the electrical activity of jaw muscles. The authors focused on three key muscles involved in jaw movement (the anterior temporal, the masseter, and the anterior belly of the digastric) by recording their activity while participants performed speech tasks. By applying the principles of network morphology, they showed that these subtle disturbances in the flow of communication between jaw muscles created a map of how these muscles coordinate with each other.
A recent study renovates surface electromyography by automatically recording and interpreting the electrical activity of jaw muscles. The authors focused on three key muscles involved in jaw movement (the anterior temporal, the masseter, and the anterior belly of the digastric) by recording their activity while participants performed speech tasks. By applying the principles of network morphology, they showed that these subtle disturbances in the flow of communication between jaw muscles created a map of how these muscles coordinate with each other.
The study looked at three groups: ALS patients with overt bulbar symptoms, those in an earlier “prodromal” phase without overt symptoms, and healthy controls. Using sophisticated graph-based analysis, the researchers examined how muscle connectivity differed between these groups and whether these changes could serve as early warning signs of bulbar dysfunction.
The results were striking. Even for prodromal patients, this method highlights subtle disruptions in the flow of communication between jaw muscles. As the disease progressed, some features of the network continued to deteriorate, while others appeared to stagnate. Crucially, these network features correlated with measurable changes in jaw movement, reinforcing the idea that ALS changes not only the strength of muscles but also how they work together.
To further their findings, the researchers used machine learning models, and training algorithms to distinguish ALS patients from healthy individuals based solely on their muscle networks. The models performed well, suggesting that the technique could one day serve as an objective, data-driven tool to diagnose ALS earlier and more accurately than current methods.
However, moving this research from the lab to clinical practice will require several steps. Larger, more diverse studies are needed to confirm that the method reliably identifies ALS while distinguishing it from other speech-impairing conditions. To be widely used, the method must also be accepted by practitioners, who like many specialists are gradually being replaced by more reliable tools. To avoid being devalued, they will need to shift their service offerings toward greater personalization and the ability to interpret not raw results, but more sophisticated information.
Nevertheless, the study represents a significant step forward. By listening more closely to the hidden rhythms of muscle coordination, researchers have found a new way to detect muscle changes in ALS, which could help clinicians stay ahead of a disease that so often eludes early diagnosis.
 They measured somatic repeat expansion over time in individual neurons from donors of different ages. They found that early-phase expansions (e.g., from 40 to 80 CAG repeats) were slow and stochastic, taking decades, while later expansions (e.g., from 80 to 150 repeats) occurred more rapidly.
They then analyzed genetic markers and DNA repair mechanisms associated with repeat instability, such as those involving DNA mismatch repair (MMR) proteins (e.g., MSH3, PMS1). Variants in these genes have been shown to influence the rate of somatic instability. The progression of CAG repeat expansion is driven by errors in DNA replication, repair, and maintenance, particularly in neurons. Key mechanisms include:
They measured somatic repeat expansion over time in individual neurons from donors of different ages. They found that early-phase expansions (e.g., from 40 to 80 CAG repeats) were slow and stochastic, taking decades, while later expansions (e.g., from 80 to 150 repeats) occurred more rapidly.
They then analyzed genetic markers and DNA repair mechanisms associated with repeat instability, such as those involving DNA mismatch repair (MMR) proteins (e.g., MSH3, PMS1). Variants in these genes have been shown to influence the rate of somatic instability. The progression of CAG repeat expansion is driven by errors in DNA replication, repair, and maintenance, particularly in neurons. Key mechanisms include: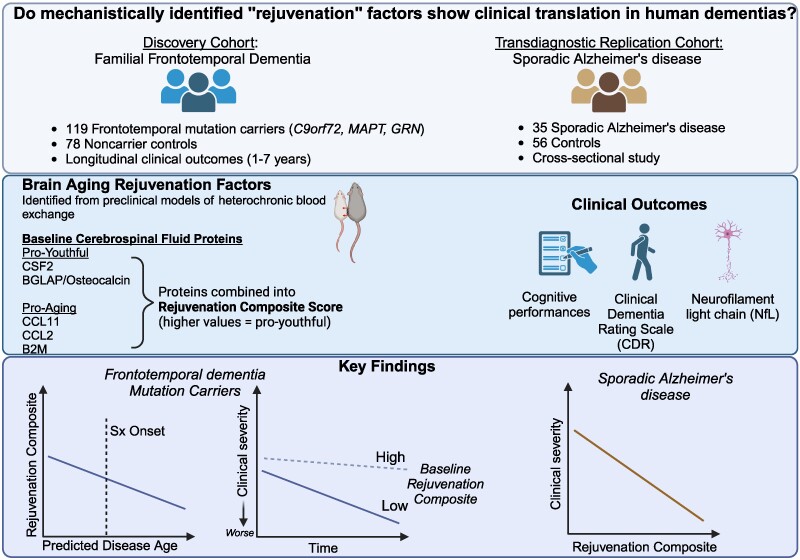 The results appear relatively reliable because the scientists found similar effects in two different types of dementia. The effects were seen across multiple measures (cognitive, functional, and biological markers).
The results appear relatively reliable because the scientists found similar effects in two different types of dementia. The effects were seen across multiple measures (cognitive, functional, and biological markers). The endoplasmic reticulum (ER) is an important organelle in cells that is involved in protein conformation. This step occurs after protein synthesis by ribosomes and after conformation, the new protein will be sent to its final destination by the Golgi apparatus. Protein conformation requires energy, so when disease occurs, the ER may not be able to properly conform the new proteins.
The endoplasmic reticulum (ER) is an important organelle in cells that is involved in protein conformation. This step occurs after protein synthesis by ribosomes and after conformation, the new protein will be sent to its final destination by the Golgi apparatus. Protein conformation requires energy, so when disease occurs, the ER may not be able to properly conform the new proteins.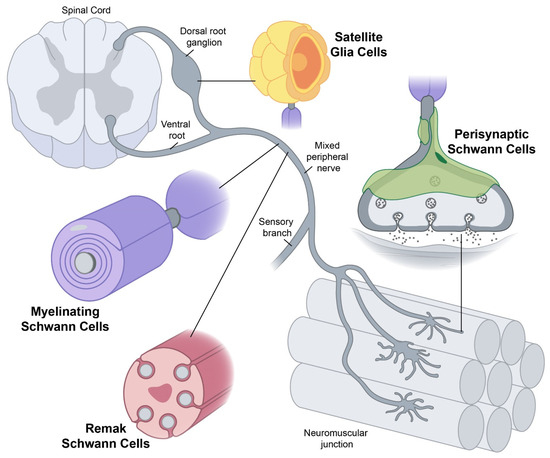 Yet conceptually associating Schwann cells and ALS is not common, ALS is a disease of the central nervous system (upper motor neurons in the brain and spine) while Schwann cells are located in the peripheral nervous system (lower motor neurons with their bodies from the spine and terminating in muscles). This does not mean there are no relations between the two types of cells, and it's a common view now that neurons are not independent, self-sufficient entities and that they are cared for by a large number of other cell types.
Yet conceptually associating Schwann cells and ALS is not common, ALS is a disease of the central nervous system (upper motor neurons in the brain and spine) while Schwann cells are located in the peripheral nervous system (lower motor neurons with their bodies from the spine and terminating in muscles). This does not mean there are no relations between the two types of cells, and it's a common view now that neurons are not independent, self-sufficient entities and that they are cared for by a large number of other cell types.