2023 was marked by two events that I find regrettable: The marketing authorization of a drug (lecanemab) after an almost unsuccessful phase III clinical trial and dangerous side effects, and the proposal to redefine the disease of Alzheimer's disease based on molecules that are probably not biomarkers of this disease to facilitate obtaining marketing authorization.
Fortunately, there are more disinterested researchers, who are working on other hypotheses on the causes of Alzheimer's disease than those of amyloid plaques. The text discussed here, by Jennifer Erichsen and Suzanne Craft particularly highlights the link between insulin sensitivity, metabolic dysregulation, and inflammatory processes in the context of Alzheimer's disease.
It is well known that people with diabetes are at greater risk (1.6 times) of decline in cognitive function. The prevalence of moderate cognitive impairment in patients with diabetes is high (45%). This is presumably because, unlike most organs, brain functions require a constant supply of glucose as an energy source, so the brain is more sensitive to abnormalities in glucose metabolism.
The role of insulin is to trigger an intracellular signal that regulates the entry of glucose into our cells. Insulin resistance is one of the characteristics of diabetes but also of neurodegenerative diseases, as well as aging. It de facto leads to a sort of brain starvation.
There is a clear link between insulin and amyloid plaques: insulin is also involved in the clearance of beta-amyloid, a protein that forms the plaques characteristic of Alzheimer's disease.
 The authors believe that an impaired blood-brain barrier allows immune cells from the body to pass through, which leads to the activation of microglia in the central nervous system. Scientists point out that immune processes intensively consume energy, therefore glucose, and therefore insulin resistance slows down immune processes.
The authors believe that an impaired blood-brain barrier allows immune cells from the body to pass through, which leads to the activation of microglia in the central nervous system. Scientists point out that immune processes intensively consume energy, therefore glucose, and therefore insulin resistance slows down immune processes.
The progression of insoluble tau to neurofibrillary tangle pathology correlates with the progression of Alzheimer's disease symptoms. Insulin metabolism has been closely linked to tau protein. Pathological accumulation of tau leads to brain insulin resistance.
The main suggestion of the authors is to combine therapeutic interventions of different natures and to minimize side effects. This is certainly an important reflection which nevertheless does not seem to be common.
They cite for example that insulin delivery with specialized devices can quickly and directly transport insulin to the central nervous system, bypassing the peripheral nervous system to avoid hypoglycemia and other adverse systemic effects. A phase II clinical trial has shown some effectiveness with a specific device.
They also cite SGLT2 inhibitors, a class of drugs commonly used in diabetes, which reduce the risk of dementia by 42% in people with type 2 diabetes.
In conclusion, researchers believe that the elimination of amyloid is insufficient to stop, much less reverse the course of Alzheimer's disease and that significant risks accompany it.
They therefore propose researching adjuvants to improve efficacy and safety. They hope this next promising and essential step in the therapeutic pathway for Alzheimer's disease will begin quickly.
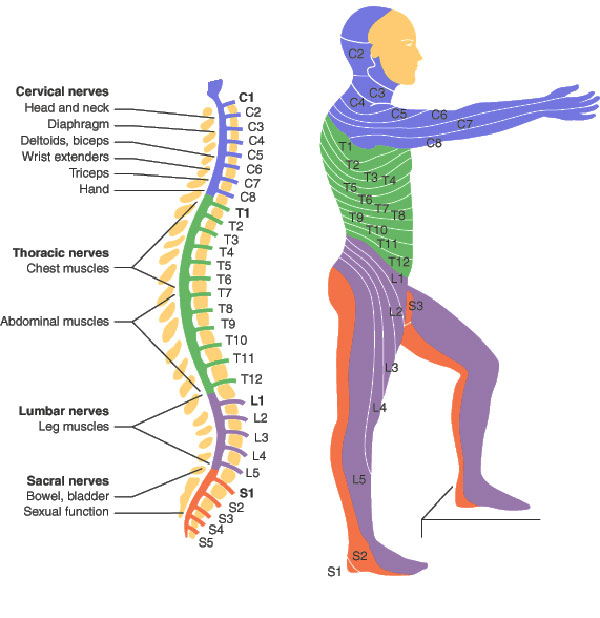 Obviously, a spinal cord injury will sever the link between the brain motor area, some upper motor neurons, and corresponding lower motor neurons and muscles. Yet it does not stop there, which is particularly interesting when we have ALS in mind. In spinal-onset ALS the disease starts in a very localized muscular for example a muscle in the thumb, and it spreads, often until respiratory muscles fail.
Obviously, a spinal cord injury will sever the link between the brain motor area, some upper motor neurons, and corresponding lower motor neurons and muscles. Yet it does not stop there, which is particularly interesting when we have ALS in mind. In spinal-onset ALS the disease starts in a very localized muscular for example a muscle in the thumb, and it spreads, often until respiratory muscles fail. .
(source:
.
(source:  Nos précédentes publications sur ce site, avertissaient déjà que cette révision aurait pour conséquence principale que les essais cliniques de médicaments seraient majoritairement approuvés, alors que la totalité des essais cliniques (324 de phase III) sur la maladie d’Alzheimer (y compris les médicaments récemment autorisés) se sont soldés par des échecs, et parfois par des effets secondaires dramatiques (ARIA).
Nos précédentes publications sur ce site, avertissaient déjà que cette révision aurait pour conséquence principale que les essais cliniques de médicaments seraient majoritairement approuvés, alors que la totalité des essais cliniques (324 de phase III) sur la maladie d’Alzheimer (y compris les médicaments récemment autorisés) se sont soldés par des échecs, et parfois par des effets secondaires dramatiques (ARIA).
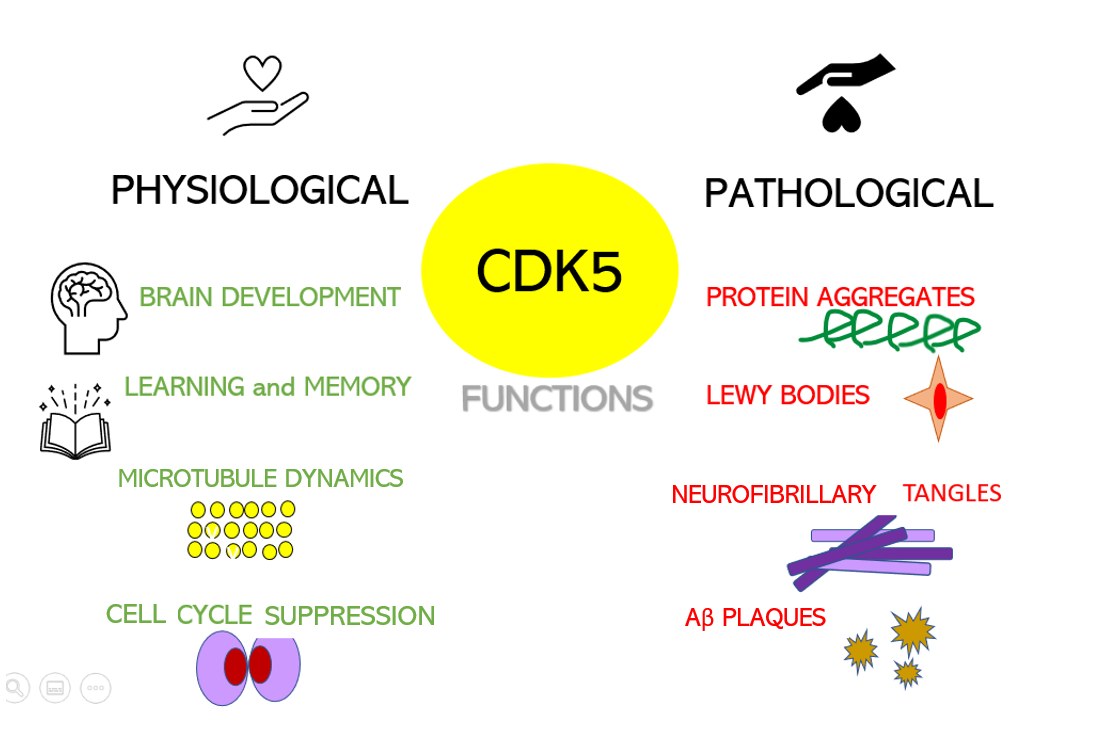 Dysregulation of CDKs, particularly cyclin-dependent kinase 5 (Cdk5), is seen in many neurological disorders, including Alzheimer's disease (AD) and Parkinson's disease (PD). Cdk5 is a unique member of the CDK family because it does not play a critical role in cell cycle progression and is not activated by a cyclin. Instead, Cdk5 is normally activated by the regulatory protein p25. Cdk5/p35/p25 activity is normally an important regulator of the proper development of the mammalian central nervous system.
Dysregulation of CDKs, particularly cyclin-dependent kinase 5 (Cdk5), is seen in many neurological disorders, including Alzheimer's disease (AD) and Parkinson's disease (PD). Cdk5 is a unique member of the CDK family because it does not play a critical role in cell cycle progression and is not activated by a cyclin. Instead, Cdk5 is normally activated by the regulatory protein p25. Cdk5/p35/p25 activity is normally an important regulator of the proper development of the mammalian central nervous system.