SUMO (Small Ubiquitin-like Modifier) proteins are a family of small proteins that are attached to and detached from other proteins in cells to modify their function. This process is called SUMOylation. SUMOylation is to signal to other cellular mechanisms that the protein attached must be processed. There are at least 4 SUMO isoforms in humans; SUMO-1, SUMO-2, SUMO-3, and SUMO-4. SUMO proteins are involved in a variety of cellular processes, such as nuclear transport, transcriptional regulation, apoptosis, and protein stability.
 Transactive response DNA-binding protein 43 (TDP-43) is a nuclear RNA binding protein (RBP) involved in RNA metabolism.
TDP-43 has a high propensity to aggregate because of its low solubility in cells and in vitro.
The aggregation propensity of TDP-43 is increased by ALS/FTD-linked mutations and upon exposure to stress and has been observed in patients with C9orf72 hexanucleotide repeat expansion, the most common genetic cause of sporadic and familial.
Transactive response DNA-binding protein 43 (TDP-43) is a nuclear RNA binding protein (RBP) involved in RNA metabolism.
TDP-43 has a high propensity to aggregate because of its low solubility in cells and in vitro.
The aggregation propensity of TDP-43 is increased by ALS/FTD-linked mutations and upon exposure to stress and has been observed in patients with C9orf72 hexanucleotide repeat expansion, the most common genetic cause of sporadic and familial.
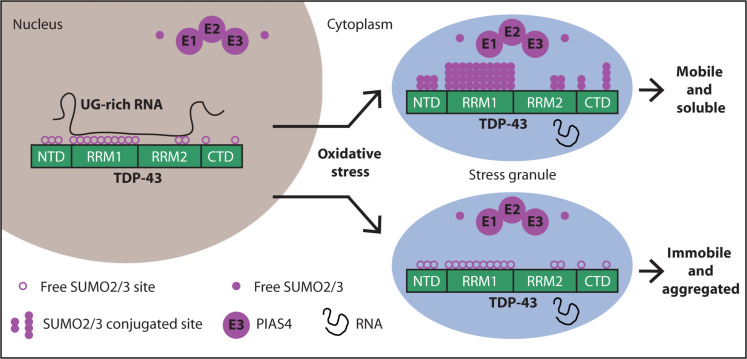
Stress conditions trigger the accumulation of TDP-43 in cytosolic stress granules. The role of stress granules in modulating TDP-43 aggregation is ambiguous. Functionally, SUMO2/3-ylation has been shown to maintain stress granules in a dynamic state: SUMOylation inhibition impairs stress granule disassembly, but the underlying mechanism is still unknown.
In this paper, the authors report that upon oxidative stress (induced by sodium arsenite), TDP-43 becomes modified by SUMO2/3 protein chains and moves from the nucleus to cytoplasmic stress granules. This conclusion is probably also valid for other stress conditions, so this study is of great interest. When researchers blocked SUMOylation, TDP-43 became less mobile within the cell, stress granules took longer to disassemble, and TDP-43 was more likely to form insoluble aggregates. Yet this is not a study on humans, the authors used various immortal cell lines, motor neuron cells, and Caenorhabditis elegans worms. Furthermore, the authors used genetic therapies to infect cells to express or repress PIAS4. This is easy to do in vitro but most probably hard to achieve in a seriously ill patient.
Because TDP-43 aggregation is central to familial and sporadic ALS, approaches aimed at preventing TDP-43 aggregation hold promise for future treatments. How cells control TDP-43 aggregation is poorly understood. Modifiers of TDP-43 solubility include molecular chaperones and posttranslational modifications. Besides ubiquitination, which is a key posttranslational modification required to clear aggregation-prone proteins, phosphorylation of TDP-43 is emerging as a protective response to counteract its misfolding. Phosphorylation of TDP-43 decreases its assembly into condensates and suppresses TDP-43 aggregation and toxicity.
Under normal growth conditions, cells prefer to modify proteins with SUMO1, while during cellular stress, SUMO2 and SUMO3 are usually conjugated in the form of SUMO2/3 chains (referred to as SUMO2/3-ylation). TDP-43 when SUMO1-ylated stays in the nucleus, does not aggregate in the cytoplasm, and its splicing activity is modified.
Using experiments in cells, the authors show that conjugation of TDP-43 with SUMO2/3 coincides with stress granule assembly. Pharmacological inhibition of TDP-43 SUMO2/3-ylation triggers TDP-43 aggregation inside stress granules.
E3 SUMO-protein ligase PIAS4 is one of several protein inhibitors of activated STAT (PIAS) proteins. PIAS proteins act as transcriptional co-regulators with at least 60 different proteins to either activate or repress transcription. The transcription factors STAT, NF-κB, p73, and p53 are among the many proteins that PIAS interacts with. PIAS4 has been shown to recruit proteins to the site of the DNA damage and promote repair.
The authors found that PIAS4 helps attach SUMO2/3 to TDP-43. - PIAS4-mediated SUMO2/3-ylation increases the solubility of TDP-43 and prevents its aggregation in the cytoplasm. - Depleting PIAS4 leads to TDP-43 aggregation In motor neurons from the human spinal cord in familial ALS cases with TDP-43 and C9orf72 mutations, reduced cytoplasmic PIAS4 correlates with increased TDP-43 aggregates.
RNA binding appears to compete with SUMOylation: When cells are not subjected to stress, TDP-43 is mainly localized inside the nucleus, binding with high affinity to RNA. When TDP-43 is bound to RNA, it's less likely to be SUMOylated. When cellular RNA levels are low, there's increased SUMO2/3 modification This suggests SUMOylation may be a protective mechanism when TDP-43 isn't bound to RNA
The authors conclude that modification with SUMO2/3 chains maintains the solubility of RNA-free TDP-43 during stress.
There are many studies on reducing TDP-43 aggregates in ALS, but this one looks much more sophisticated than the previous ones. Yet this is mostly an in-vitro study. Long pre-clinical studies must be conducted on mammals to verify if a simple and safe agonist of PIAS4 (which does not exist today) could improve the health of ALS patients.
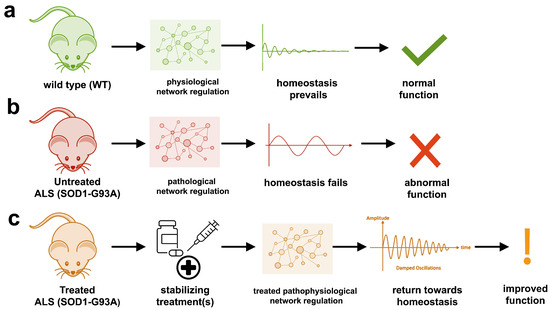 The study uses an innovative and integrative framework to model the regulatory dynamics of wild-type (WT) and SOD1-G93A ALS mice. The models are based on first-order ordinary differential equations (ODEs) that describe how the system output evolves over time. The research uses dynamic meta-analysis to synthesize experimental data from the literature and parameter optimization based on genetic algorithms to infer missing data. Indeed, to build a model, data are needed and here these are obtained from results reported in the literature on SOD1-G93A ALS mouse models.
The study uses an innovative and integrative framework to model the regulatory dynamics of wild-type (WT) and SOD1-G93A ALS mice. The models are based on first-order ordinary differential equations (ODEs) that describe how the system output evolves over time. The research uses dynamic meta-analysis to synthesize experimental data from the literature and parameter optimization based on genetic algorithms to infer missing data. Indeed, to build a model, data are needed and here these are obtained from results reported in the literature on SOD1-G93A ALS mouse models.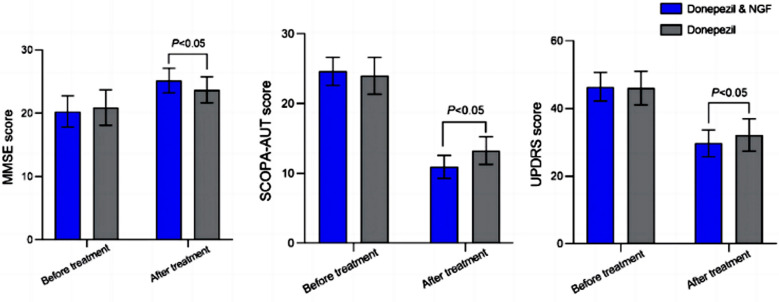 The authors focused on adiponectin, an adipocytokine, i.e. a molecule produced by adipose tissue, which is involved, among other things, in the regulation of lipid and glucose metabolism. Adiponectin modulates inflammatory cascades by modifying the action and production of inflammatory cytokines, but the link between adiponectin and Parkinson's disease is not obvious unless we consider that Parkinson's disease is due to a metabolic disorder. The relationship with the soluble tumor necrosis factor receptor (sTNFR) is even less obvious. Nothing in the article explains why these two molecules were studied.
The authors focused on adiponectin, an adipocytokine, i.e. a molecule produced by adipose tissue, which is involved, among other things, in the regulation of lipid and glucose metabolism. Adiponectin modulates inflammatory cascades by modifying the action and production of inflammatory cytokines, but the link between adiponectin and Parkinson's disease is not obvious unless we consider that Parkinson's disease is due to a metabolic disorder. The relationship with the soluble tumor necrosis factor receptor (sTNFR) is even less obvious. Nothing in the article explains why these two molecules were studied. Alzheimer's disease research has produced many hypotheses over the years, including cholinergic, inflammatory, viral, mitochondrial, tau, and amyloid. However, none of these hypotheses have led to treatments that can stop or reverse the disease. This leads to a search for new theories to explain these failures. But this may be because interventions occur too late in the disease progression, with brain damage irreparable and compensatory mechanisms saturated.
Alzheimer's disease research has produced many hypotheses over the years, including cholinergic, inflammatory, viral, mitochondrial, tau, and amyloid. However, none of these hypotheses have led to treatments that can stop or reverse the disease. This leads to a search for new theories to explain these failures. But this may be because interventions occur too late in the disease progression, with brain damage irreparable and compensatory mechanisms saturated.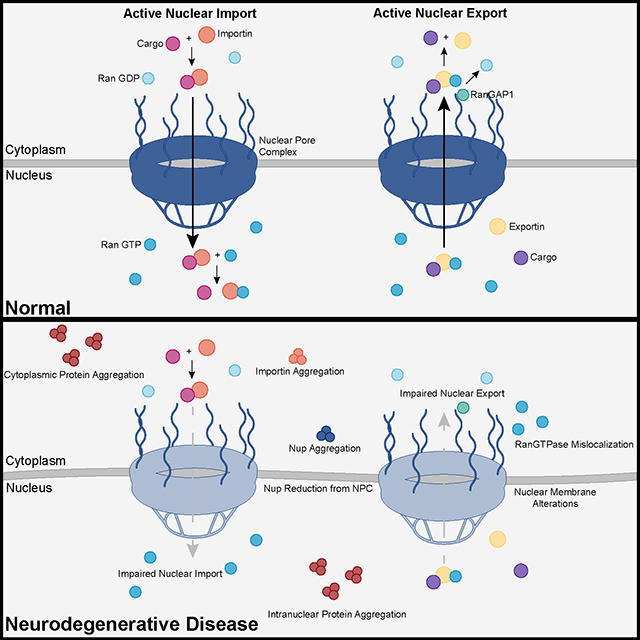 Recent advances into the underlying pathogenic mechanisms have associated mislocalization and aberrant accumulation of disease-related proteins with defective nucleocytoplasmic transport and its mediators called karyopherins.
Recent advances into the underlying pathogenic mechanisms have associated mislocalization and aberrant accumulation of disease-related proteins with defective nucleocytoplasmic transport and its mediators called karyopherins.