Le texte ci-dessous est dérivé de l'article suivant qui est très intéressant, mais présente de nombreuses modifications de mon fait. Ces modifications relativisent l'optimisme pour les techniques scientifiques et le relative oubli de l’aspect humain dans ce genre de littérature académique. Mais encore une fois il s'agit d'un excellent article très informatif sur l'état de l'art des ASO.
Thérapies basées sur les ASO
Les oligonucléotides antisens (ASO) sont de courtes molécules d'ADN ou d'ARN conçues pour se lier à des régions spécifiques de l'ARNm cible et donc interférer avec la production de protéine dans les cellules infectées. La production de protéine est indispensable à la vie de la cellule, les protéines sont à la fois utilisées comme matériaux et comme moyen de coordination entre les différentes organelles cellulaires. Les ASO peuvent moduler l’épissage pré-ARNm, augmenter les niveaux de protéines fonctionnelles et diminuer les niveaux de protéines toxiques.
 Ces dernières années, l’utilisation des ASO a donné un nouvel élan à la recherche et au développement de thérapies efficaces pour de nombreuses maladies auparavant incurables comme la SBMA, l'atrophie musculaire spinale (SMA) et la sclérose latérale amyotrophique (SLA).
Dans ce domaine, le recours aux ASO a conduit à de grands succès et à de grands échecs.
Ces dernières années, l’utilisation des ASO a donné un nouvel élan à la recherche et au développement de thérapies efficaces pour de nombreuses maladies auparavant incurables comme la SBMA, l'atrophie musculaire spinale (SMA) et la sclérose latérale amyotrophique (SLA).
Dans ce domaine, le recours aux ASO a conduit à de grands succès et à de grands échecs.
Le plus grand succès a certainement été le développement d'une thérapie pour la SMA basée sur un ASO appelé nusinersen, capable de compenser le manque de protéine SMN dans le gène SMN1. Le médicament améliore considérablement la fonction motrice et augmente la survie des patients atteints de SMA. En quelques années seulement, il a été possible de passer de la phase préclinique à l'approbation et à la commercialisation du médicament grâce à une collaboration efficace entre les différentes parties prenantes.
Un autre résultat très positif a été obtenu dans le traitement de la SLA associée à des mutations du gène SOD1. Tofersen est un ASO conçu pour le traitement des patients SLA porteurs d’une certaine mutation du gène SOD1. Les données recueillies jusqu'à présent ont montré qu'il peut ralentir la progression de la maladie chez certains malades, surtout si le traitement est commencé tôt après l'apparition des symptômes.
D'autres études cliniques à long terme sont toujours en cours, mais la FDA a approuvé le tofersen en 2023 dans le cadre de son programme d'approbation accélérée, et l'approbation de l'EMA est arrivée en février. 2024. Il s’agit d’une étape importante dans l’histoire de la recherche sur la SLA qui a montré pour la première fois que la maladie, au moins sous certaines de ses formes (concernant malheureusement moins de 2% des patients), peut être traitable.
Ces résultats ont stimulé la recherche de thérapies basées sur l'ASO pour la SLA associée à des mutations dans d'autres gènes. Des essais cliniques avec des ASO ciblant les mutations des gènes FUS, STMN2 et ATXN2 sont en cours et montreront dans les années à venir si ces thérapies sont efficaces.
Le cas de l’expansion du gène C9orf72 mérite une discussion à part. Compte tenu de la fréquence de cette mutation dans les formes familiales et sporadiques de la SLA, des ASO dirigés contre l’ARNm sens, ciblant l’ARNm contenant l’expansion répétée, ont été développés et testés avec des résultats encourageants dans des modèles animaux. Sur la base de données prometteuses, deux sociétés différentes ont lancé des essais cliniques de phase 1/2 avec des ASO ciblant différentes régions de l'ARNm de répétition. Malheureusement, les deux essais ont été interrompus après une analyse intermédiaire des résultats, car ni les critères d'évaluation principaux ni secondaires n'étaient atteints.
À ce stade, on ne sait pas exactement quelle pourrait être la cause de ces échecs bien que diverses hypothèses ont été suggérées.
Aspects généraux importants du développement de thérapies basées sur l'ASO.
Les scientifiques classent l'effet des ASO en deux principaux mécanismes pathologiques provoqués par des variantes pathogènes : la perte de fonction et le gain de fonction. Cette classification semble pourtant rudimentaire, la plupart des mécanismes biologiques étant extrêmement complexes. En général, les variantes perte de fonction sont relativement plus faciles à gérer que les variantes gain de fonction. Les gènes perte de fonction sont souvent approchés en restaurant des productions de protéines saines, tandis que les gènes gain de fonction sont approchés en limitant la production ou en augmentant la dégradation de protéines altérées.
La perte de fonction
L'approche thérapeutique générale de la perte de fonction dans le cas de la SMA, bénéficie de l'existence du gène SMN2 qui produit aussi de la protéine SMN (Le gène SMN2 est dérivé du gène SMN1 au cours de l'évolution), car il était possible de cibler un site de SMN2 et ainsi d'augmenter la synthèse d'une protéine SMN fonctionnelle complète.
Le gain de fonction
Les gains de fonction incluent, les variantes faux-sens et les répétitions étendues, et ils confèrent des fonctions supplémentaires, inopportunes, à la protéine, augmentant sa propension à former des agrégats, peut-être car la cellule ne sait pas gérer ou dégrader cette production de protéines incongrues. Par conséquent, une façon de développer une thérapie efficace consiste à dégrader les agrégats toxiques ou à empêcher leur formation. Il existe deux options différentes.
Une stratégie sélective implique l’utilisation d’ASO qui dégradent la variante toxique (mutante) tout en préservant la forme normale de la protéine. Encore faut-il que les cellules du patient produisent suffisamment de protéine normale.
D’un autre côté, un ciblage du gène affecté pourrait réduire à la fois les variantes toxiques et saines. Il s’agit d’une approche plus simple et ne nécessite pas l’utilisation de thérapies adaptées (différents ASO pour différentes mutations), mais cela implique que les fonctions physiologiquement nécessaires de la protéine de type sain sont également réduites. En effet l’inactivation non spécifique d’une protéine a forcément des conséquences graves et imprévisibles.
Il est ridicule de penser que nous puissions avoir des mécanismes biologiques qui seraient inutiles, et c'est assez incroyable que l'on envisage cette méthode pour soigner des humains. Ces effets négatifs sur la protéine de type sain sont probablement à l’origine de l’échec du traitement par ASO chez les patients SLA porteurs de la mutation du gène C9orf72. De tels effets négatifs n’ont cependant pas été observés avec le traitement par tofersen chez les patients SLA présentant des mutations SOD1. Cependant, un suivi à long terme de ces patients est nécessaire.
Un exemple de cette stratégie de blocage de production, est la tentative de correction des défauts d’épissage dus à l’épuisement nucléaire de la protéine TDP-43. Un des nombreux rôles de cette dernière protéine est de corriger les défauts lors de la transcription de protéines. Or les cellules produisant en permanence environ 20 000 types de protéines différents malgré les différents stress auxquels elles sont confrontées (surtout chez les patients âgés), les erreurs de productions sont nombreuses et nécessitent de nombreux mécanismes de correction.
Le principal avantage thérapeutique de cette stratégie est qu’une population beaucoup plus large de patients SLA que les seuls porteurs de mutations pourrait bénéficier de cette stratégie thérapeutique, car une mauvaise localisation de TDP-43 est observée chez presque tous les patients SLA. D'ailleurs cette mauvaise localisation de TDP-43 est observée dans d'autres maladies neurodégénératives comme la FTD ou la maladie d'Alzheimer. Mais cette stratégie, compte tenu du rôle fondamental de TDP-43, serait équivalente à une forme d'empoisonnement du patient, bien cela n'empêche pas de nombreux scientifiques et laboratoires de la considérer comme voie thérapeutique.
Une approche plus raisonnable consiste à tenter de corriger les défauts d’épissage d’ARNm spécifiques. Il n’est pas clair si la correction de ces anomalies d’épissage uniques et spécifiques s’avérera efficace. Il est possible que plusieurs de ces ARNm mal épissés doivent être corrigés avec un cocktail d’ASO avant d’obtenir un bénéfice thérapeutique.
Alternativement, il peut être utile d'identifier d'autres entités biologiques jouant un rôle clé dans le processus d'épissage médié par TDP-43. SYF2 est un facteur d'épissage pré-ARNm qui est recruté dans le spliceosome pour réguler l'épissage. Lorsqu'il est régulé négativement, il inverse la pathologie du TDP-43 et améliore la fonction du TDP-43, y compris le traitement de l'ARN, dans les modèles précliniques. Ainsi, la régulation négative de SYF2 médiée par ASO pourrait restaurer un mauvais épissage de plusieurs ARNm mais aussi perturber l’épissage de protéines saines. Mais là encore il est illusoire de penser qu'inhiber une protéine puisse avoir des effets bénéfiques. Pour savoir si cette stratégie fonctionne également chez les patients atteints de SLA, les essais cliniques seront cruciaux.
Un mode d'administration contestable
Actuellement, les ASO sont administrés par administration intrathécale, une procédure plutôt invasive et techniquement exigeante. Quelques cas de malades réagissant particulièrement bien ont été récemment médiatisés par des laboratoires universitaires et pharmaceutiques. Ce qui n'est pas dit c'est que ces malades subissent une chirurgie à risque chaque semaine. Bien que couramment utilisée en milieu clinique, le caractère invasif et le coût de la procédure stimulent le développement de technologies d’administration alternatives. Du point de vue du malade c'est un risque à courir, mais ce n'est sûrement pas tenable sur la durée et c'est un coût phénoménal pour les sécurité sociales.
La voie intrathécale est une procédure invasive qui peut être remplacée par le développement d’ASO conjugués à des nanoparticules.
Des progrès en chimie visant à utiliser de nouveaux adjuvants sont en cours de développement. L'utilisation de particules d'administration, telles que des nanoporteurs polymères enrobés de glucose et des ASO conjugués à des peptides, est très prometteuse pour l'avenir, car elles peuvent traverser la barrière hémato-encéphalique et améliorer le transport des ASO dans le système nerveux.
Cependant, la délivrance par nanoparticules peut présenter une toxicité liée à la nature des nanoparticules utilisées. Par exemple, les nanoparticules à base de protéines peuvent exercer une cardiotoxicité, et une hépatotoxicité importante.
Traitement chez les personnes asymptomatiques
Les traitements chez les personnes asymptomatiques sont toujours l'objet de controverses éthiques, qui peut prédire qui va être malade de la SLA? Pourquoi condamner des personnes apparemment saines à un traitement toute leur vie? Plus prosaïquement comment convaincre les systèmes de sécurité sociale et mutuelles qu'économiquement ces traitements à vie ont un intérêt, à l'heure où on réduit l'assistance aux patients atteints de maladies chroniques?
Ce qui motive ces propositions c'est qu'au moment où les premiers symptômes de la maladie apparaissent, les motoneurones et plus globalement le système musculaire ont déjà subi des dommages importants qui ne peuvent être éliminés. Par conséquent, le traitement ne peut que ralentir ou, au mieux, arrêter la progression de la maladie. Deux essais cliniques chez des individus pré-symptomatiques avec un diagnostic génétique de SMA (NURTURE, NCT023865539) et porteurs de mutations du gène SOD1 (ATLAS, NCT04856982) sont en cours sur cette question. Si les résultats sont positifs, nous pourrions assister à un changement révolutionnaire pour certaines maladies du système moteur, qui vont passer du statut d’incurables et mortelles à celui de traitables.
Dans le cas de la SMA, le dépistage néonatal (NBS) permet d'initier immédiatement un traitement spécifique pour les enfants atteints de SMA afin de stopper la perte irréversible des motoneurones et la progression de la maladie et d'assurer un développement moteur comme celui des enfants sans maladie neuromusculaire. Dans le cas de la SLA, étant donné que les mutations du gène SOD1 représentent environ 20 % des maladies d'origine familiales et jusqu'à 2 % des cas sporadiques, un dépistage rapide des mutations SOD1 devrait être effectué chez tous les nouveaux patients SLA présentant des présentations à la fois familiales et sporadiques. Mais il y a des dizaines de mutations différentes de SOD1, certaines foudroyantes, d'autres relativement lentes à tel point que le patient décède seulement à un âge avancé.
Conclusion
En résumé, la thérapie ASO a fait des progrès remarquables ces dernières années, apportant des bénéfices significatifs au traitement des maladies des motoneurones. Le plus grand succès a été le développement du nusinersen, le premier traitement efficace contre la SMA approuvé par la FDA et l'EMA, capable d'améliorer les symptômes et de ralentir la progression de la maladie. Cela a été suivi quelques années plus tard par le tofersen, qui a été approuvé pour traiter les patients SLA présentant l'une des mutations SOD1. En revanche, le chemin à parcourir est encore long concernant d’autres formes de SLA associées à des mutations d’autres gènes, notamment C9orf72. Une compréhension plus approfondie des mécanismes pathogénétiques liés à la présence de mutations, ainsi que le développement de molécules de plus en plus efficaces et performantes, pourraient permettre de développer de nouvelles thérapies contre ces maladies neurodégénératives.
 Gyromitra esculenta
Gyromitra esculenta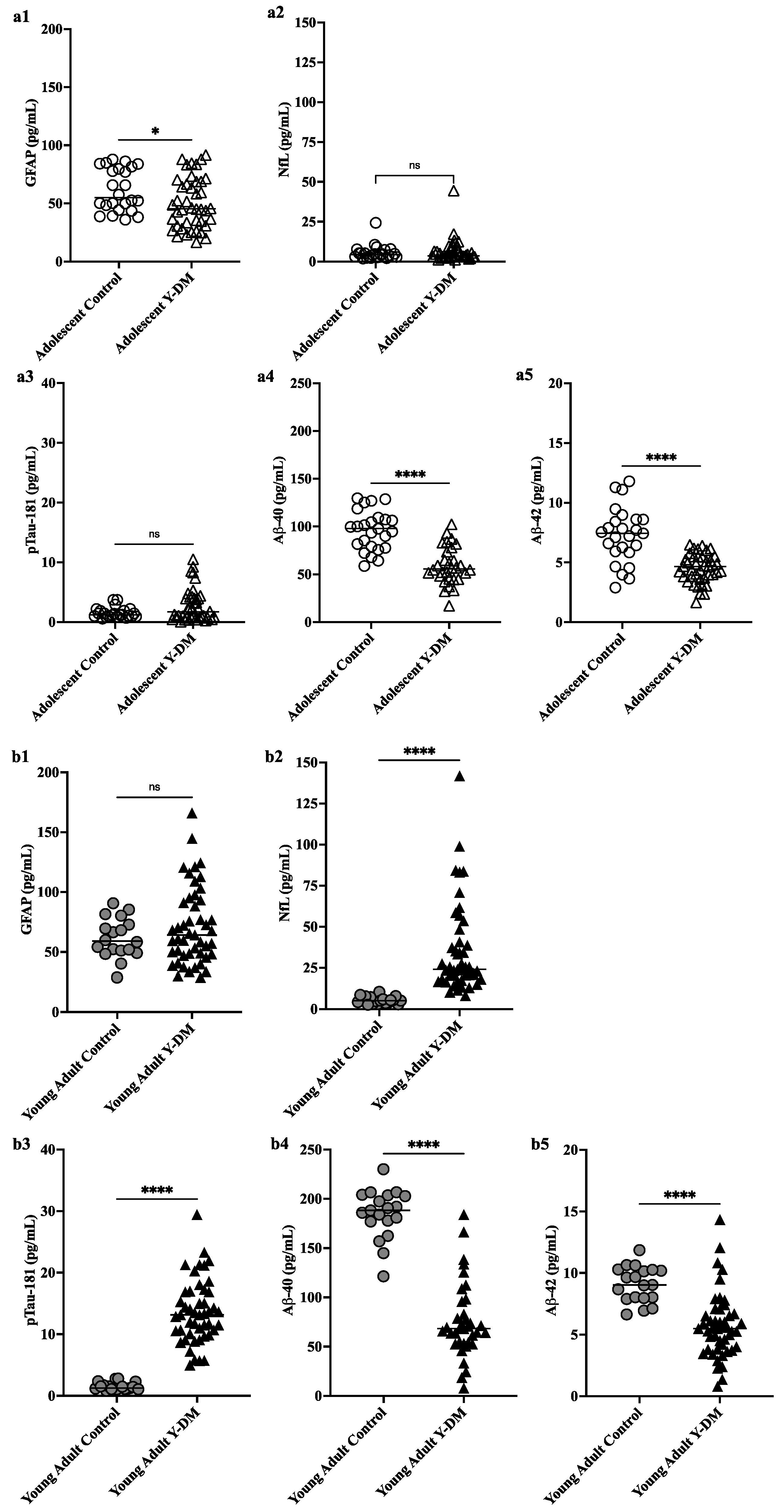 This study aimed to explore neurodegenerative disease biomarkers in cohort-derived biomarker banks as changes in key plasma biomarkers between the time of diabetes diagnosis and early adulthood have been correlated with worsening cognitive function in young adults with early-onset diabetes.
This study aimed to explore neurodegenerative disease biomarkers in cohort-derived biomarker banks as changes in key plasma biomarkers between the time of diabetes diagnosis and early adulthood have been correlated with worsening cognitive function in young adults with early-onset diabetes. Sleep efficiency in older adults. Sleep deprivation increases amyloid-β (Aβ) concentrations in the interstitial fluid of experimental animal models and in cerebrospinal fluid in humans, while increased sleep decreases Aβ. Sleep abnormalities may therefore represent a risk factor for neurodegeneration.
Sleep efficiency in older adults. Sleep deprivation increases amyloid-β (Aβ) concentrations in the interstitial fluid of experimental animal models and in cerebrospinal fluid in humans, while increased sleep decreases Aβ. Sleep abnormalities may therefore represent a risk factor for neurodegeneration.