Alors que le but de ce blog est habituellement de donner des nouvelles des laboratoires sur les maladies neurodégénératives, ce post est différent: Il s'agit d'une tentative de simplification du langage, de traduction et de condensation d'un long article publiant l'opinion d'un certain nombre de professionnels de la recherche sur la SLA sur les animaux modèles.
Nous avons trouvé cet article passionnant mais très, très long. Espérons que ce post sera plus facile à lire.
Plus de 500 essais cliniques sur la SLA ont été entrepris depuis une trentaine d’années. Une revue récente a établi qu'entre 2008 et 2019, 125 essais testant 76 médicaments chez 15 000 patients SLA ont été réalisés, presque tous sans identifier de traitement efficace.
Nous ne pouvons pas passer aux essais cliniques sans passer par des modèles animaux pour évaluer la vaste gamme de paramètres requis pour le développement de médicaments.
La SLA est compliquée -- en partie parce qu'il ne s'agit pas d'une seule maladie
La sclérose latérale amyotrophique (SLA) est une maladie neurodégénérative dévastatrice, progressive et incurable qui frappe généralement vers la cinquantaine. Elle a été nommé ainsi par le neurologue Charcot, mais en France on l'appelle couramment "la maladie de Charcot". Les motoneurones sont les cellules principalement affectées dans la SLA, mais la maladie se situe sur un spectre de démence frontotemporale, et il est clair que d'autres types de cellules que les motoneurones y sont impliqués.
L'hétérogénéité de la maladie se manifeste dans l'observation courante que certains patients atteignent le stade terminal de la maladie un an après dès l'apparition des symptômes, alors que d’autres progressent lentement sur plus d'une décennie, par exemple Stephen William Hawking a vécu pendant 55 ans avec la SLA.
Dans presque tous les cas de la maladie de Charcot, les motoneurones portent des inclusions cytoplasmiques de la protéine TDP-43. Cela se produit indépendamment du fait que la maladie soit une SLA « sporadique », c’est à dire ne provenant d'aucune cause connue, ou qu'elle soit provoquée par des mutations parmi les nombreux gènes qui ont été associés à la SLA familiale.
Absence de traitements efficaces
Presque 150 ans après l'identification de cette maladie, nous n'avons toujours pas d'image unifiée du mécanisme pathologique ni de traitements efficaces. Il est probable que de nombreux mécanismes peuvent contribuent à la mort des motoneurones.
Quatre traitements pour la SLA: Le riluzole, l'édaravone, l'AMX0035 et le Tofersen sont actuellement approuvés aux USA, mais ces médicaments ne prolongent la vie que de quelques mois et peuvent ne pas convenir à tous les patients.
De nouveaux traitements sont explorés qui offrent de l'espoir aux patients atteints de formes génétiques de la SLA, y compris SOD1-ALS, FUS-ALS et potentiellement C9orf72, qui représente jusqu'à 40 % des cas familiaux.
Pourtant ces stratégies thérapeutiques ne ciblent qu'une seule des différentes voies biologiques impliquées dans les processus pathologiques au sein de chaque sous-type de la maladie de Charcot.
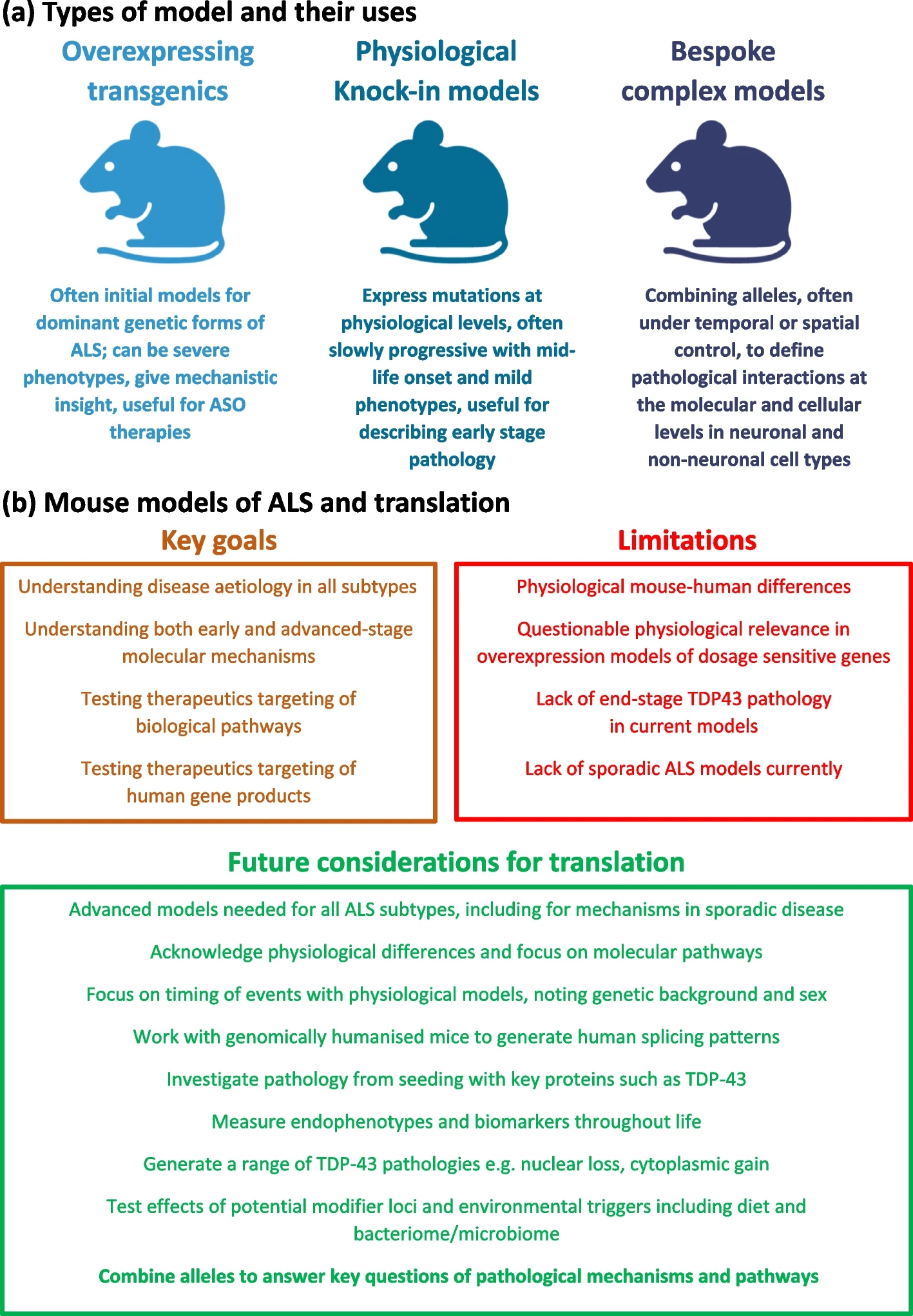
Il n'y a pas de modèle unique pour les modèles de souris de la SLA.
L'un des plus grands défis actuels de la recherche sur la SLA est que même si nous pouvons générer des modèles approximatifs de la maladie sur des souris de laboratoire, il est toujours difficile de créer des modèles animaux reflétant vraiment la SLA, ce qui handicape le développement de médicaments.
Le premier gène SLA découvert a été SOD1 en 1993, à l’époque les scientifiques pensaient que la découverte d’un médicament était proche, et cette découverte a été suivie en 1994 par la création du premier modèle de souris SLA transgénique. Les modèles de souris TARDBP et FUS n'ont été publiés que plus de dix ans plus tard. En conséquence, pendant longtemps, les animaux transgéniques surexprimant SOD1 ont été les seuls modèles disponibles de la SLA, puis par la suite, les moins chers.
Les souches de souris ayant un gène SOD1 transmuté développe généralement une maladie rapide même chez les jeunes souris, ce qui n’est pas le cas chez l’humain, car la SLA est habituellement une maladie liée à l’âge.
Les patients dotés d’un gène SOD1 dysfonctionnel (on devrait dire ‘allèle’) représentent moins de 2 % des cas de la maladie de Charcot humaine et semble maintenant être une aberration possible dans le paysage complexe de la SLA, car les tissu de patients ayant un gène SOD1 ou FUS dysfonctionnel ne montrent pas de dépôts de la protéine TDP-43 qui est observée dans la plupart des cas de la maladie de Charcot. Il en est de même pour les malades ayant une mutation du gène FUS-ALS. Ainsi, les thérapies qui ont été développées et testées sur des souris transgéniques SOD1 ou FUS peuvent ne pas être applicables à la plupart des patients.
Difficultés à modéliser la maladie
Les allèles dysfonctionnels des gènes TARDBP et FUS se sont révélés plus difficiles à modéliser car ces gènes sont sensibles au dosage (c'est-à-dire au nombre de copies de ces gènes dans le génome), et les souris transgéniques surexpriment généralement leur transgène (le gène implanté artificiellement pour simuler la maladie) à cause de des méthodes, relativement agressives, qui sont utilisées pour introduire le transgène dans le génome des souris.
Par exemple, même une faible surexpression de la protéine TDP-43 (non mutée) chez une souris transgénique entraîne chez celle-ci une paralysie des membres postérieurs!
De plus, les gènes originaux de la souris restent intacts et continuent à produire des protéines fonctionnelles, ce qui signifie que la perte de fonction d'un gène ne peuvent pas être correctement modélisés chez les souris transgéniques conventionnelles à moins qu'elle ne soit couplée à une inactivation du gène natif (knock-out).
Une meilleure stratégie pour concevoir des modèles TARDBP-ALS et FUS-ALS consiste à générer des animaux exprimant une dose «physiologique» normale à partir d'une seule copie du gène. Il y a ainsi plusieurs souches de souris actuellement disponibles qui sont porteuses de mutations dans les gènes endogènes de la SLA.
Par exemple, parmi les modèles endogènes Tardbp (du nom du gène qui génère la protéine TDP-43), celui de la mutation M323K qui induit lorsqu'elle est homozygote une mort progressive des motoneurones.
La plupart des cellules d’animaux et de plantes ont pour chaque gène, un "locus" (aussi appelé allèle) sur chacun des deux chromosomes, on dit qu'ils sont diploïdes. Si les deux allèles d’un gène sur chacun des deux chromosomes sont identiques, on dit que ce gène est homozygote. S'ils sont différents, l'organisme dit hétérozygote.
Curieusement les différentes mutations du gène Tardbp induisent des phénotypes très éloignés les uns des autres: Ainsi la mutation Q331K induit un dysfonctionnement cognitif, alors que les mutations homozygotes M337V et G298S affectent les jonctions neuromusculaires et produisent une gliose médullaire à des stades tardifs, sans neurodégénérescence franche, etc.
Ces modèles de souris knock-in résolvent le problème de la sensibilité au dosage, mais ne s'attaquent pas aux différences subtiles entre les gènes humains et murins.
Introduction de gènes humains dans le génome des animaux modèles
De nouveaux modèles dans lesquels le gène natif de la souris est remplacé par la séquence correspondante chez l'humain (on dit orthologue) ont donc été développés, notamment dans les modèles actuels de FUS-ALS.
L’une des étapes de la production de protéine à partir d’un gène s’appelle l’épissage. l’épissage est un processus par lequel les ARN transcrits à partir de l'ADN génomique peuvent subir des étapes de découpage et réassemblage avant d'être envoyés aux ribosomes. Les segments conservés s’appellent des exons et ceux qui sont éliminés s’appellent des introns.
Une telle humanisation génomique se traduit généralement par des épissage similaires à ceux observés chez les humains, qui sont généralement plus complexes que ceux observés chez les souris.
Les animaux modèles ayant un gène humain peuvent fournir de nouvelles informations, par exemple, ces modèles semblent assez représentatifs des changements précoces observés chez les patients atteints de FUS-ALS.
*** Inadéquation entre le temps de la recherche et les contraintes apportées par les modèles animaux***
Comme la maladie se développe seulement au milieu de la vie d'une souris, à environ 12 mois, ces nouveaux modèles très complexes sont lents à produire des résultats et donc coûteux à utiliser pour le développement de médicaments.
Pour cette raison, souvent ils ne sont pas adoptés par l’industrie pharmaceutique ou les biotech. En outre, l'étude de ces souris dans le cadre d'un doctorat peut être difficile à mener compte tenu du besoin de publication qui se fait sentir dès la deuxième année.
Ainsi, il existe une tension entre la précision de ces nouveaux modèles et les contraintes des milieux universitaires et industriels. Bien sûr, cette situation n'est pas unique à la recherche sur la SLA, mais hélas elle oriente le développement de nouveaux médicaments potentiels vers des modèles animaux adaptés aux contraintes de temps et financières des expérimentateurs plutôt qu'à une modélisation fidèle de la maladie humaine.
L'absence de dépôt de TDP-43 dans les modèles de souris Tardbp/TARDBP
Etre capable de récapituler le dépôt de TDP-43 chez la souris serait d'une grande utilité pour modéliser toute la gamme de la pathologie SLA car les inclusions cytoplasmiques pathologiques de FUS, TDP-43 et SOD1 caractérisent différents sous-types humains de la maladie de Charcot.
Cependant, le dépôt de TDP-43 n'est pas spécifique de la SLA, car il a été trouvé dans d'autres troubles tels que la maladie d'Alzheimer, les maladies polyglutamines, l'encéphalopathie TDP-43 liée à l'âge à prédominance limbique (LATE) et à la suite d'atteintes neurologiques, y compris l'exposition au méthylmercure et l'hypoperfusion.
Ainsi si les aggrégats de protéine TDP-43 mal repliés et inccorectement localisés dans la cellule sont des marqueurs de a SLA, il n'est pas clair s'ils sont la cause de la SLA, ou une conséquence du développement de la maladie. Des approches pour modifier les modèles de souris SLA pour produire un dépôt de TDP-43 sont à l'étude.
Il est clair que chaque espèce est différente aussi nous ne devrions pas nous attendre à une "validité totale" des modèles de souris : la physiologie, la durée de vie, la taille et les antécédents génétiques d'une souris auront un impact sur la façon dont une mutation de la maladie se manifestera.
Nous fabriquons des modèles de souris aussi pour comprendre certaines voies moléculaires et cellulaires spécifiques qui résultent de la maladie, et non pour imiter exactement la condition humaine.
Différences anatomiques significatives entre rongeurs primates
Il y a des différences anatomiques significatives dans le système moteur des rongeurs et des primates, par exemple dans le tractus corticospinal. Le tractus corticospinal est une voie nerveuse allant du cortex cérébral à la moelle épinière qui est responsable des mouvements volontaires des membres et du tronc qui sont singulièrement atteints lors de la SLA. C'est cette voie nerveuse qui comprend les motoneurone supérieur et inférieurs, ainsi que des interneurones ou des liaisons directes entre les deux types de motoneurones.
Les différences anatomiques entre les souris et les humains ont des implications pour la modélisation de la SLA chez la souris, car le diagnostic de la SLA est basé sur la présence à la fois de signes de motoneurone supérieur (UMN) et de LMN, et nous ne pouvons pas modéliser exactement la dégénérescence UMN du tractus corticospinal.
Chez les primates, le tractus corticospinal descend dans les segments ventraux et latéraux de la moelle épinière, il est alors composé de motoneurones supérieurs, et forme des connexions monosynaptiques avec les motoneurones inférieurs (LMN). Les axones des motoneurones inférieurs quittent la moelle épinière par la racine ventrale pour former les nerfs périphériques, qui innervent la musculature du corps.
En revanche, chez les rongeurs, le tractus corticospinal ne descend que dans la moelle épinière dorsale et, contrairement aux primates, les fibres du tractus corticospinal ne forment pas de connexions directes avec les motoneurones spinaux. Pourtant ces connexions neuronales cortico-motrices directes que l'on trouve chez les primates semblent être particulièrement impliquées au début de la SLA.
Travailler avec des modèles de souris complexes - leçons tirées de la recherche sur le cancer
La présence de nombreux sous-types de maladies et le faible nombre de patients de la SLA a probablement fait que nous avons peut-être développé des médicaments qui ont fonctionné pour certaines personnes, mais que cela n’a pas été mis en évidence sur le plan statistique parce que dans l’essai clinique ces patients ne représentaient qu’une minorité. L'utilisation de modèles complexes est devenue la norme dans la recherche sur le cancer, car cette stratégie peut offrir des avancées majeures dans la compréhension de la pathologie et fournir de nouvelles informations inattendues.
Grâce à la recherche sur le cancer, nous avons les outils pour répondre aux questions difficiles de la recherche sur la SLA, notamment en créant des modèles animaux spécialement conçus pour étudier toute la complexité de la situation in vivo, dans chaque cellule au cours de la vie, en tenant compte de la génétique, de l'environnement, de l'âge de la cellule et de l'animal entier.
Stratification des patients et besoin de plus de modèles pour comprendre les phénotypes de la SLA
La SLA possède au fil des malades, une variabilité importante dans le temps et le site d'apparition de la maladie, les pathologies moléculaires, les taux de progression et bien sûr la survie. Cette hétérogénéité de la maladie sous-tend l'un des principaux facteurs limitant les progrès des essais cliniques sur la SLA : le défi de la stratification des patients.
La comparaison de données entre les modèles de souris et les patients doit être stratifiée par les sous-types de la maladie de Charcot, et au moyen d'une gamme de biomarqueurs comparables dans les deux espèces. Idéalement, les paramètres mesurables reflétant une meilleure qualité de vie chez les patients atteints de la maladie de Charcot devraient inclure une fonction musculaire accrue ainsi que des biomarqueurs fiables surveillant la progression altérée de la maladie. Enfin, la comparaison des similitudes et des différences de pathologie dans chaque sous-type de la maladie de Charcot, humain et dans les modèles animaux, est essentielle.
Ainsi, il est important de prendre en compte dans la conception des essais la classification a priori des sous-types de patients en fonction des modèles pronostiques et des biomarqueurs disponibles.
En plus d'identifier les patients susceptibles d'avoir une évolution plus rapide de la maladie, il est essentiel d'identifier les 10 à 20 % des patients SLA qui ont une survie supérieure à 10 ans . L'étude de ces deux sous-ensembles de patients devraient nous permettre d'obtenir des informations fondamentales sur le développement de la maladie et sur les effets des thérapies qui sont expérimentées.
La répartition au hasard des patients dans les deux branches (traitement et placébo) doit également tenir compte d’autres facteurs, mais une limitation de cette approche par stratification des malades est que les essais cliniques de la SLA sont forcément effectués sur un nombre très limité de malades. Les essais cliniques de la SLA flirtent déjà avec la limite de signification statistique, stratifier les patients ferait perdre toute signification aux éventuelles conclusions issues de ces essais. Après tout il s’agit d’une maladie rare.
Une thérapie développée pour une pathologie spécifique, telle qu'un dysfonctionnement de la protéine de liaison à l'ARN (TDP-43), peut ne pas fonctionner dans une autre forme de la SLA causée par exemple par la mutation d'une protéine de trafic membranaire, d'où la nécessité aussi d'une stratification des patients avec des données de mécanique cellulaire.
Bien que nous ayons des modèles génétiques de souris qui donnent une variabilité phénotypique, nous n'avons pas encore de modèles pour différentes régions anatomiques humaines d'apparition de la maladie. En effet si les scientifiques étudient assidument les motoneurones supérieurs dans le cadre de la SLA, la maladie débute de façon focale (par exemple au niveau du pouce) ce qui semble impliquer les jonctions neuromusculaires entre motoneurone inférieure et muscle. Le point le plus frappant pour un patient de la SLA, ce n'est pas l'incapacité totale à ce servir de ses muscles, celà n'arrive que tardivement dans le développement de la maladie, c'est plutôt la fonte musculaire intense et l'absence de fiabilité des mouvements volontaires.
Des modèles complexes tels que les mutations inductibles spécifiques aux tissus, par exemple, devraient aider, car nous avons également besoin des principales voies et événements biochimiques responsables de ces différences tissulaires humaines.
Les médicaments contre la SLA que nous avons peut-être manqués
Un résultat tragique des obstacles résultant de la présence de nombreux sous-types de maladies et du faible nombre de patients est que nous avons peut-être déjà développé des médicaments qui ont fonctionné pour certaines personnes, mais celà n'a pas été étudié parce que les conclusions sur essais cliniques ne sont élaborées que si elles concernent des nombres significatifs, en général plusieurs centaines de patients.
Plusieurs composés se sont révélés efficaces chez les souris transgéniques SOD1 et pourraient bien s'être avérés bénéfiques chez les patients SOD1-ALS .
Un exemple est l'arimoclomol, un co-inducteur de la protéine de choc thermique, qui s'était bien comporté dans les études précliniques de souris transgéniques SOD1-ALS et qui a été soumis à un essai en double aveugle et contrôlé par placebo chez 38 patients atteints de SOD1-ALS à progression rapide, dont 19 ont reçu un placebo, 17 ont reçu de l'arimoclomol.
L'étude clinique de phase II a échoué à atteindre ses buts statistiques, mais a montré que l'Arimoclomol était relativement bien toléré et que le traitement avait une certaine efficacité. En particulier le sous-groupe des participants ayant une mutation SOD1 A4V a semblé avoir un effet thérapeutique certain. Cependant le faible nombre de participants n’a pas permis de dégager une indication claire au niveau statistique. Il n’y a donc pas eu d’étude de phase III.
Comme l'ont indiqué les auteurs, la petite taille de l'échantillon reflétait la rareté de la population SOD1-ALS qui est estimée à seulement 320 patients au total sur l'ensemble des États-Unis, tous n'auraient pas consenti, et le recrutement pour cette étude sur l'Arimoclomol était alors en compétition avec deux autres essais cliniques. Bien que > 10 % des patients atteints de SOD1-ALS aient été recrutés, ce nombre était trop faible pour passer à l'essai de phase III prévu chez les patients SOD-ALS.
Conclusion:
Ainsi, les difficultés de stratification des patients, y compris le petit nombre de patients pour la plupart des sous-types de la maladie de Charcot, sont un problème clé pour la conception de médicaments, les nouveaux modèles animaux complexes pourraient être une réponse à ce challenge.
La recherche sur la SLA a besoin de ces systèmes modèles sophistiqués pour disséquer les pathologies et développer des traitements conventionnels, génétiques et/ou cellulaires pour chacunes des nombreuses formes de cette maladie.
 This condition affects individuals, mostly adults over the age of 50, who physically act out their dreams during sleep, resulting in injuries to themselves or their bed partners. It's also suspected to be involved in premisses of Parkinson's disease. The study, published in the Journal of Neuroscience, presents a novel model that characterizes how REM sleep behavior disorder develops due to neurodegeneration associated with the accumulation of tau protein.
This condition affects individuals, mostly adults over the age of 50, who physically act out their dreams during sleep, resulting in injuries to themselves or their bed partners. It's also suspected to be involved in premisses of Parkinson's disease. The study, published in the Journal of Neuroscience, presents a novel model that characterizes how REM sleep behavior disorder develops due to neurodegeneration associated with the accumulation of tau protein. The disease known as ALS is multifaceted, in fact not only are there believed to be dozens of ALS subtypes, but there are also dozens of diseases that have similar symptoms to ALS. This greatly complicates not only the diagnosis, which is always long and uncertain, but also the clinical trials which are based on the implicit assumption that all the patients treated have the same disease, which is probably not the case for the trials of drugs for degenerative diseases.
The disease known as ALS is multifaceted, in fact not only are there believed to be dozens of ALS subtypes, but there are also dozens of diseases that have similar symptoms to ALS. This greatly complicates not only the diagnosis, which is always long and uncertain, but also the clinical trials which are based on the implicit assumption that all the patients treated have the same disease, which is probably not the case for the trials of drugs for degenerative diseases. Il me semble donc que cette activité commerciale se prêterait bien à une innovation disruptrice, par exemple en confiant à des infirmiers "senior" comme les
Il me semble donc que cette activité commerciale se prêterait bien à une innovation disruptrice, par exemple en confiant à des infirmiers "senior" comme les  Parmi ces sociétés on trouve Google qui depuis longtemps cible le monde médical.
Parmi ces sociétés on trouve Google qui depuis longtemps cible le monde médical. 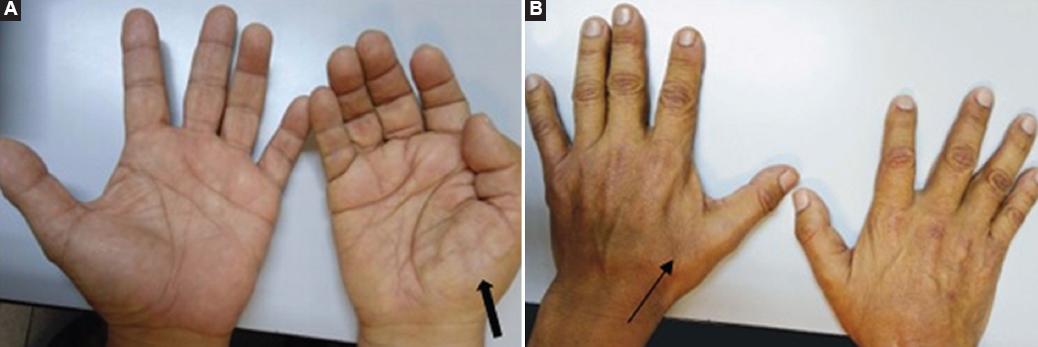 Hand surgeons are commonly the first providers to evaluate patients with undiagnosed ALS (the famous split-hand syndrome). So in early phases, the symptoms of amyotrophic lateral sclerosis (ALS) can mimic those of compressive neuropathies, such as carpal and cubital tunnel syndromes, especially early in a patient's clinical course. Scientists surveyed members of the American Society for Surgery of the Hand and found that 11% of active and retired members have performed nerve decompression surgeries on patients later diagnosed with ALS. Other scientists found that in 95% of cases, the diagnostic errors are, related to physician cognitive error that results from a lack of knowledge about the disease and inadequate decisions
Hand surgeons are commonly the first providers to evaluate patients with undiagnosed ALS (the famous split-hand syndrome). So in early phases, the symptoms of amyotrophic lateral sclerosis (ALS) can mimic those of compressive neuropathies, such as carpal and cubital tunnel syndromes, especially early in a patient's clinical course. Scientists surveyed members of the American Society for Surgery of the Hand and found that 11% of active and retired members have performed nerve decompression surgeries on patients later diagnosed with ALS. Other scientists found that in 95% of cases, the diagnostic errors are, related to physician cognitive error that results from a lack of knowledge about the disease and inadequate decisions The primary function of mitochondria in normal cells is to employ oxidative phosphorylation of ADP via the electron transport chain to produce ATP. Compared to non-excitable cells such as fibroblasts (a standard experimental platform for studying mitochondrial dynamics), neurons are especially dependent upon mitochondrial ATP because of the high energy requirements for electrochemical neurotransmission.
The primary function of mitochondria in normal cells is to employ oxidative phosphorylation of ADP via the electron transport chain to produce ATP. Compared to non-excitable cells such as fibroblasts (a standard experimental platform for studying mitochondrial dynamics), neurons are especially dependent upon mitochondrial ATP because of the high energy requirements for electrochemical neurotransmission.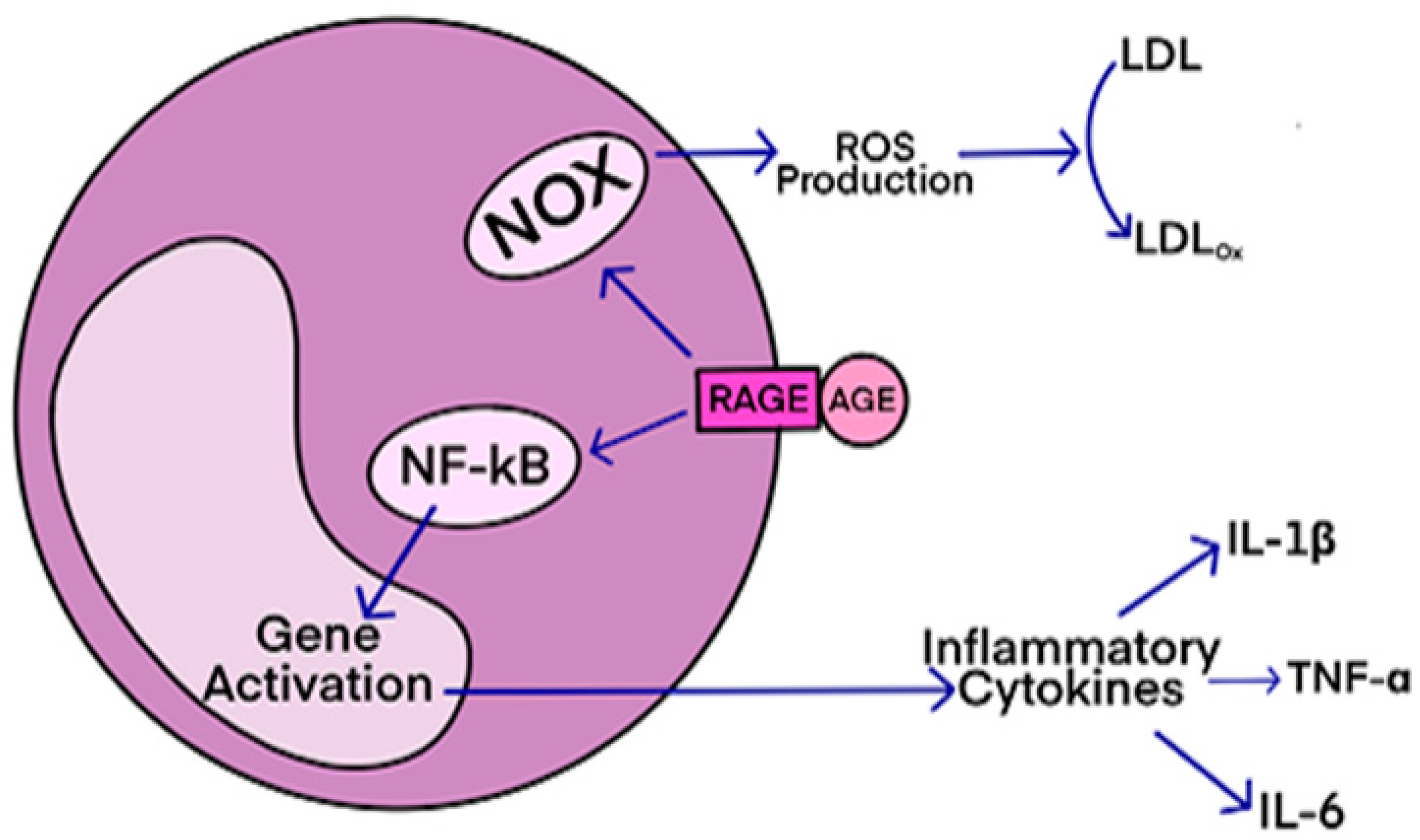 AGEs have diverse structures, but only a limited number have been characterized. Some AGEs are small molecules formed through proteolytic degradation of protein-crosslinked or protein-modified AGEs. Imbalance between the formation and destruction of AGEs, particularly under conditions of oxidative stress, results in excessive accumulation and disease progression.
AGEs have diverse structures, but only a limited number have been characterized. Some AGEs are small molecules formed through proteolytic degradation of protein-crosslinked or protein-modified AGEs. Imbalance between the formation and destruction of AGEs, particularly under conditions of oxidative stress, results in excessive accumulation and disease progression.

 The European Interdisciplinary Council on Aging hosted a 2-day virtual meeting on November 24-25, 2022, to review the state of knowledge on the link between infection and neurological disorders, and dementia. We describe in this post
The European Interdisciplinary Council on Aging hosted a 2-day virtual meeting on November 24-25, 2022, to review the state of knowledge on the link between infection and neurological disorders, and dementia. We describe in this post