Early nonmotor symptoms in Lewy body disease
Parkinson's disease is a progressive neurodegenerative movement disorder with symptoms of rest tremor, muscular rigidity, slowness of movement, postural impairment, and later on often dementia which is sometimes attributed to drugs. Many people not diagnosed with Parkinson's have parkinsonism and others have only Lewy body dementia.
This medical characterization parallels ALS which has many mimics and shares a strong genetic and molecular background with fronto-temporal dementia. There is also a distinction between “body-first” and “brain-first” disease in ALS and Parkinson's disease. Both diseases mostly affect older people and the prevalence of Parkinson's disease is expected to continue to increase with the aging of the population in most developed countries.
Pathologically, Parkinson's disease is characterized by intraneuronal Lewy body inclusions prominently containing misfolded α-synuclein, which normally functions as an intracellular trafficking protein, as well as prominent neuronal death of dopaminergic neurons in the midbrain substantia nigra pars compacta. This disrupts dopamine neurotransmitter production and signaling in the basal ganglia circuit, a deep, central part of the brain.
While clinical findings of Parkinson's disease and Lewy body dementia are prominently associated with aberrant α-synuclein deposits in the central nervous system, pathologic α-synuclein can also be found in the peripheral nervous system. It has been postulated that central α-synucleinopathies may begin in the peripheral nervous system before spreading to the central nervous system.
Common early nonmotor symptoms in these disorders, such as orthostatic hypotension, constipation, and erectile dysfunction in men prefigure impairment of the peripheral and autonomic nervous systems rather than impairment of the CNS (the brain and spinal cord). Anosmia or hyposmia is also an early sign of Parkinson's disease. Despite these insights about the peripheral neurons, the main focus on central nervous system pathology and related symptoms drove the field.
Yet it is difficult to assess the presence of misfolded α-synuclein in the central nervous system (CNS). An ability to identify patients at risk with low-cost and easy-to-interpret biomarkers would facilitate the testing and implementation of disease-modifying therapies.
Cardiac noradrenergic dysfunction as an early marker
There is an ongoing search for biomarkers in many chronic diseases. The well-publicized rationale is that it would help to diagnose early diseases. Indeed many neurodegenerative diseases are not diagnosed quickly, often it need several years of examinations which causes distress to patients and families. A less publicized rationale is that it would help companies quickly get market authorizations from authorities, a good example is the ongoing proposal to revise the criteria for Alzheimer's diagnosis.
Goldstein et al. report the results of the prospective, longitudinal PDRisk study (ClinicalTrials.gov NCT00775853).
The team investigated an NIH-developed positron emission tomography (PET) tracer, 18F-dopamine, to assess dysfunction of the cardiac noradrenergic system as a prelude to the development of Parkinson's disease and Lewy body dementia. Though relatively small, this study demonstrated a remarkably accurate predictive value of low cardiac uptake of 18F-dopamine for central Lewy body disease. This finding was further corroborated by cerebrospinal fluid 3,4-dihydroxyphenylacetic acid (DOPAC).
Goldstein et al. demonstrate that a combination of cardiac noradrenergic cell loss and inefficient sequestration of catecholamines in residual cardiac sympathetic nerves precedes the onset of central Lewy body diseases, at least in a population with several identified nonmotor Parkinson's disease risk factors. The precise timing of cardiac noradrenergic cell loss to central nervous system dopaminergic cell loss remains to be elucidated. As the authors acknowledge, this paradigm may be more effective in identifying preclinical disease in patients with “body-first” rather than “brain-first” central Lewy body disease (10), since the choice of nonmotor risk factors for entry into the PDRisk study probably biases toward the “body-first” paradigm.
The catecholaldehyde hypothesis
There is a hypothesis for the pathogenesis of Parkinson’s disease that centers on an accumulation of 3,4-dihydroxyphenylacetaldehyde (DOPAL) in dopaminergic neurons, this hypothesis is sometimes called the catecholaldehyde hypothesis. DOPAC is a metabolite (a by-product) of the neurotransmitter dopamine which the authors had previously demonstrated is low in individuals with preclinical Parkinson's disease. DOPAC can be oxidized by hydrogen peroxide, leading to the formation of toxic metabolites which destroy dopamine storage vesicles in the substantia nigra. This may contribute to the failure of levodopa treatment of Parkinson's disease. A MAO-B inhibitor can prevent this from happening.
 It is also possible that there is an association of Parkinson's disease with a higher risk of important cardiovascular events like stroke and myocardial infarction (MI). 18F-dopamine is not the first tracer to demonstrate in identifying cardiac sympathetic dysfunction, however, Goldstein et al. are the first to demonstrate this relationship in a longitudinal, long-term prospective study of cardiac noradrenergic imaging in individuals with specific, self-reported nonmotor risk factors for Parkinson's disease.
It is also possible that there is an association of Parkinson's disease with a higher risk of important cardiovascular events like stroke and myocardial infarction (MI). 18F-dopamine is not the first tracer to demonstrate in identifying cardiac sympathetic dysfunction, however, Goldstein et al. are the first to demonstrate this relationship in a longitudinal, long-term prospective study of cardiac noradrenergic imaging in individuals with specific, self-reported nonmotor risk factors for Parkinson's disease.
Timing of therapeutic interventions
At this time, there are no interventions that can convincingly prevent the development or progression of central Lewy body diseases. Instead, treatment for both Parkinson's disease and Lewy body dementia relies mostly on symptomatic therapies, typically replacing or augmenting the progressively declining levels of the neurotransmitter dopamine. The amount of dopamine replacement needed increases as the disease progresses, and at a certain point, it creates dreadful adverse effects.
It is estimated that 50% to 80% of dopaminergic neurons in the substantia nigra are lost by the time noticeable clinical symptoms appear in Parkinson's disease. The peripheral nervous system presents an attractive target for developing methods of detecting α-synuclein pathology early, before the onset of widespread central nervous system damage.
Future implications
The application of this longitudinal, prospective protocol, will stimulate further studies. For example, 123I-MIBG SPECT, skin and gut α-synuclein immunofluorescence, or seed amplification assays from cerebrospinal fluid or nasal secretions also have the potential to identify prodromal or early disease in at-risk individuals.
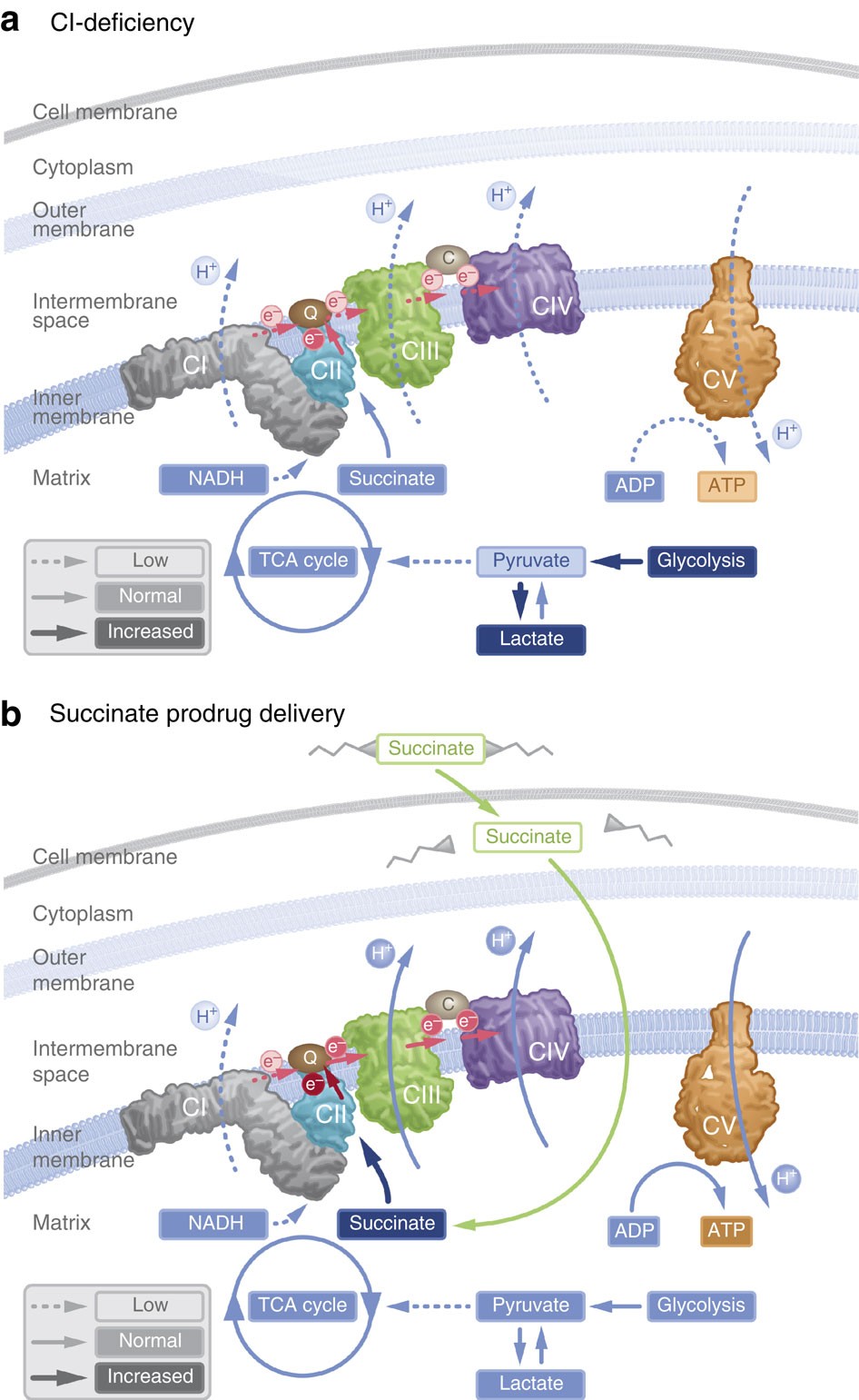 In particular, supplementation with dimethyl succinate, a substrate bypassing the inhibited step in the TCA cycle, suggests a potential therapeutic strategy to mitigate mitochondrial dysfunction in Alzheimer's disease. Dimethyl succinate (DMS), a membrane-permeant form of succinate, could serve as a pro-drug to provide substrate to the next enzymatic step in the TCA cycle, succinate dehydrogenase (SDH).
In particular, supplementation with dimethyl succinate, a substrate bypassing the inhibited step in the TCA cycle, suggests a potential therapeutic strategy to mitigate mitochondrial dysfunction in Alzheimer's disease. Dimethyl succinate (DMS), a membrane-permeant form of succinate, could serve as a pro-drug to provide substrate to the next enzymatic step in the TCA cycle, succinate dehydrogenase (SDH). Yichang Jia, Ph.D., Co-founder of SineuGene
Yichang Jia, Ph.D., Co-founder of SineuGene