Constipation and sialorrhea (impaired swallowing) have been considered to be important factors of neurodegenerative diseases (Parkinson's disease, but it is also common in ALS, Alzheimer, and autistic disorders), although the exact mechanism is still controversial.
Constipation and dementia have similar epidemiological characteristics. A study found that people with Alzheimer's disease and constipation decline nearly 3 times quicker than people with Alzheimer's disease but no constipation.
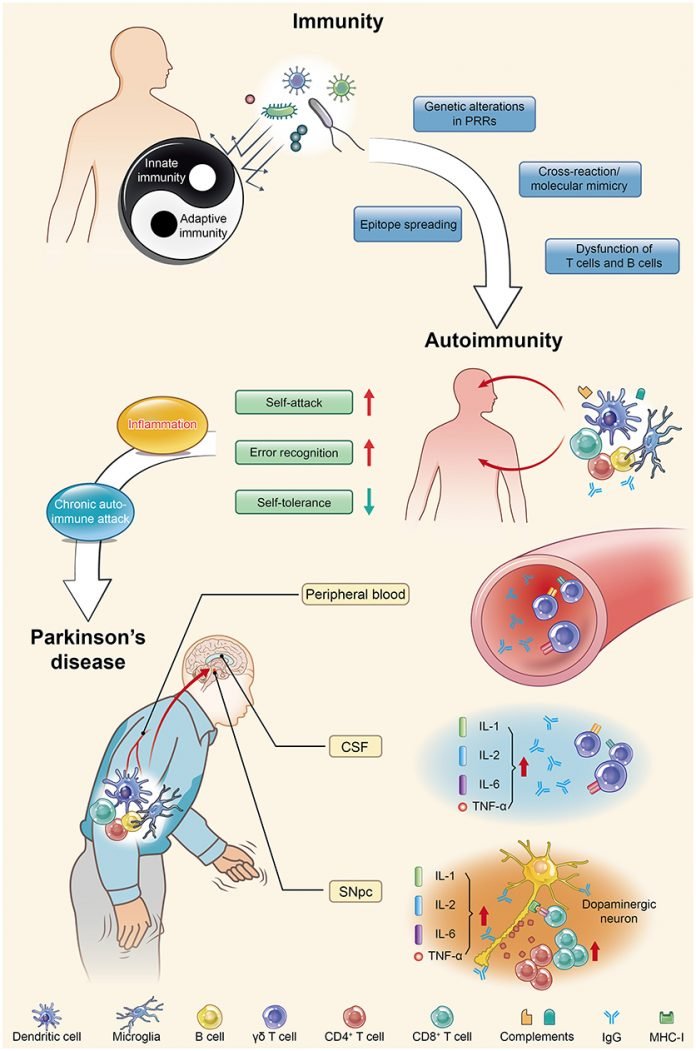 Constipation is present in approximately 70% of Parkinson's disease patients, is 3-fold more prevalent in Parkinson's disease patients than healthy controls, and occurs as early as 20 years prior to the onset of motor symptoms. Inflammatory bowel disease patients who receive anti-tumor necrosis factor alpha (TNF-α) therapy exhibit a 78%–100% reduction in Parkinson's disease incidence compared with those who do not receive such therapy.
Constipation is present in approximately 70% of Parkinson's disease patients, is 3-fold more prevalent in Parkinson's disease patients than healthy controls, and occurs as early as 20 years prior to the onset of motor symptoms. Inflammatory bowel disease patients who receive anti-tumor necrosis factor alpha (TNF-α) therapy exhibit a 78%–100% reduction in Parkinson's disease incidence compared with those who do not receive such therapy.
Constipation is often neglected by scientists and doctors who attribute it to poor hygiène, poor diet, lower water intake, comorbidities (stroke, diabetes), and brain health degradation.
In consequence, constipation symptoms are treated with pharmacologic drugs, while underlying causes are ignored and therefore not treated.
Most studies on constipation in dementia patients focus on the population with α-synucleinopathies [Parkinson’s disease dementia, dementia with Lewy bodies].
Studies have shown that total truncal vagotomy, but not selective vagotomy, was associated with a lower prevalence of Parkinson's disease which hints at propagation of the disease from the gut to the brain via the vagus nerve.
20 years ago Heiko Braak hypothesized that in Parkinson's disease, a pathogen reaching the gut initiates pathology that spreads to the CNS. The main portals for the central delivery of α-synuclein are thought to be the olfactory bulb and vagus nerve.
Peripheral inflammation is likely implicated in PD pathogenesis because high levels of pro-inflammatory cytokines (e.g., TNF-α, interleukin [IL]-1β, IL-6, and interferon [IFN]γ) are found in PD patients and correlate negatively with disease duration. This is true also in ALS,
or Alzheimer's disease.
PD patients also possess circulating T cells, mostly CD4+ subtypes, which recognize specific α-syn-derived neo-epitopes. T cells are one of the important types of white blood cells in the immune system and play a central role in the adaptive immune response. This immune response is restricted to patients carrying a specific HLA haplotype (B∗07:02 C∗07:02 DRB5∗01 DRB1∗15:01 DQA1∗01:02 DQB1∗06:02).
There are several immune cell types. White blood cells include three main subtypes; granulocytes, lymphocytes, and monocytes.
CD8+ T cells are cytotoxic lymphocytes, they kill virus-infected cells, as well as cancer cells. CD8+ T cells are also able to use small signaling proteins, known as cytokines, to recruit other types of cells when mounting an immune response.
CD4+ T cells, function as "helper cells". They activate memory B cells and cytotoxic T cells, which leads to a larger immune response. They also secrete cytokines.
- Regulatory T cells provide a mechanism of tolerance, which prevents immune cells from inappropriately reacting against their own cells. Alas, these same regulatory T cells can also be co-opted by cancer cells to prevent the recognition of, and an immune response against, tumor cells.
A different kind of lymphocyte, Memory B cells circulate in the bloodstream in a quiescent state, sometimes for decades. Their function is to memorize the characteristics of the antigen that activated their parent B cell during an initial infection such that if the memory B cell later encounters the same antigen, it triggers an accelerated and robust secondary immune response.
In each vertebrate cell, protein molecules are continually synthesized and degraded. Some small parts of degraded proteins (peptides) are sent to the surface of the cell where MHC molecules on the cell surface keep them and display them at the intention of the host's leukocytes.
These peptides are specific to one host, so if a leukocyte encounters a cell that displays peptides that are known to be from pathogens, they kill the cell. For most cell types this is not a threat to the host's health as many cell types are quickly renewed, yet this is damaging some cell types such as neurons which reproduce slowly or not at all (motorneurons).
These small peptides displayed by MHC molecules at the surface of vertebrate cells are called epitopes. MHC molecules constitute a very wide class of molecules with many types and sub-types. In humans, MHC molecules are called HLA molecules.
It's known that α-synuclein, the molecule which causes Parkinson's disease, is recognized by CD4+ T cells. One epitope, α-syn32-46 (a part of the α-synuclein molecule), binds with a strong affinity to the HLA-DRB1∗15:01 allele which is implicated in autoimmune diseases. This may not be an accident but on the contrary some anti-microbial mechanism. A parallel could be drawn with beta-amyloids in Alzheimer's disease.
 Scientists just reported that making a mouse model with leucocytes that react to a subset of α-synuclein molecule, triggers intestinal inflammation, leading to transient constipation and weight loss, yet with no detectable effects in the central nervous system (CNS). In other words, far from defending the body, the leucocyte of this mouse model are attacking their own body cells. Given that this auto-immune artificially created disease implicated a part of the molecule incriminated in Parkinson's disease and creates effects found in Parkinson's patients, it is reasonable to suspect it has some similarity with human disease.
Scientists just reported that making a mouse model with leucocytes that react to a subset of α-synuclein molecule, triggers intestinal inflammation, leading to transient constipation and weight loss, yet with no detectable effects in the central nervous system (CNS). In other words, far from defending the body, the leucocyte of this mouse model are attacking their own body cells. Given that this auto-immune artificially created disease implicated a part of the molecule incriminated in Parkinson's disease and creates effects found in Parkinson's patients, it is reasonable to suspect it has some similarity with human disease.
More precisely the scientists created a mouse strain lacking major histocompatibility complex class II (MHCII) and expressing the human HLA-DRB1∗15:01 allele. This was to model PD patients who possess circulating T cells that recognize specific α-synuclein (α-syn)-derived epitopes.
Next, the authors had to make the mice's immune system to react α-synuclein derived epitopes. They used a classic strategy which is to administrate the peptide along with some immunologic adjuvant. Vaccines commonly use such immunologic adjuvants. The authors used myelin oligodendrocyte glycoprotein peptide (MOG)35–55 plus complete Freund’s adjuvant (CFA) emulsion combined with i.v. administration of Bordetella pertussis toxin. The idea between this complex assemblage is that when leucocytes will recognize the adjuvant as hostile, CD4+ T cells will associate it with α-synuclein derived epitopes. So when CD8+ T cells will encounter cells with such epitopes, they will kill them.
This triggered intestinal inflammation and loss of enteric neurons in the submucosal plexus (SP) of the small intestine, leading to transient constipation and weight loss, with no detectable effects in the CNS. Bulk RNA sequencing (RNA-seq) of the gut reveals that α-syn32-46 immunizations induce innate and adaptive immune responses and IFN signaling. Single-cell RNA-seq (scRNA-seq) of immune cells showed altered gene signatures in CD4+ TH1 and TH17 lamina propria lymphocytes from complete Freund’s adjuvant (CFA)/α-syn32-46-immunized mice, characteristic of antigen-experienced tissue-resident memory (TRM) cells found in mucosal barriers during infection and chronic inflammation.
Depletion of CD4+, but not CD8+, T cells partially rescued enteric neurodegeneration, which confirms the hypothesized mechanism where CD4+ are responsible for immunization.
In human inflammatory bowel disease, CD4+ CD69+ CD103+ TRM cells are proposed to accumulate in the gut and trigger intestinal inflammation.57,62 A similar CD4+ TRM population (CD161+ CCR5+) produces proinflammatory cytokines in Crohn’s disease.
Thus, interactions of α-syn32-46 with the HLA-DRB1∗15:01 allele are critical for the induction of enteric features resembling those seen in prodromal Parkinson's disease, suggesting that additional hits may be required for the development of CNS symptoms.
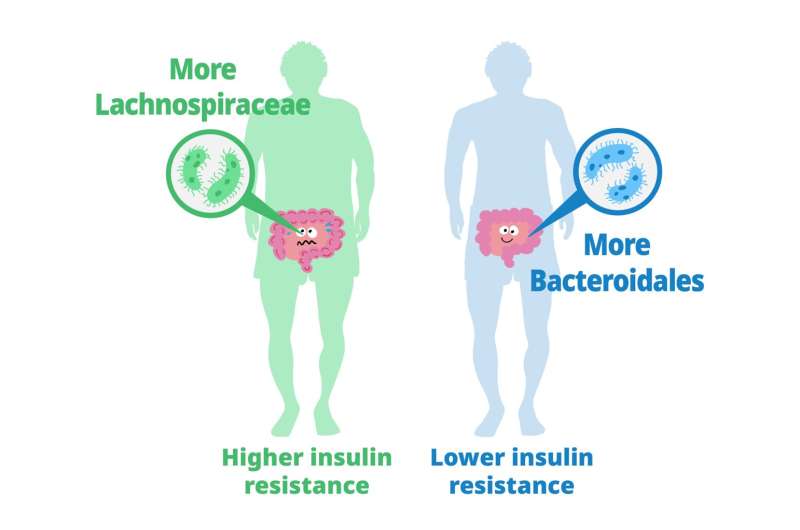 Previous studies have explored the role of gut microbiota in metabolizing nutrients in insulin resistance. This research aims to uncover the mechanisms underlying this relationship using a multi-omics approach. The study analyzes data from 306 individuals without diabetes, focusing on insulin resistance as defined by HOMA-insulin resistance scores.
Previous studies have explored the role of gut microbiota in metabolizing nutrients in insulin resistance. This research aims to uncover the mechanisms underlying this relationship using a multi-omics approach. The study analyzes data from 306 individuals without diabetes, focusing on insulin resistance as defined by HOMA-insulin resistance scores. A
A  I analyze below the first article because
I analyze below the first article because  For them, symptoms often start with weakness, unreliability, and thinning of the thumb or calf, and with time the disease progresses to other (skeletal) muscles.
For them, symptoms often start with weakness, unreliability, and thinning of the thumb or calf, and with time the disease progresses to other (skeletal) muscles.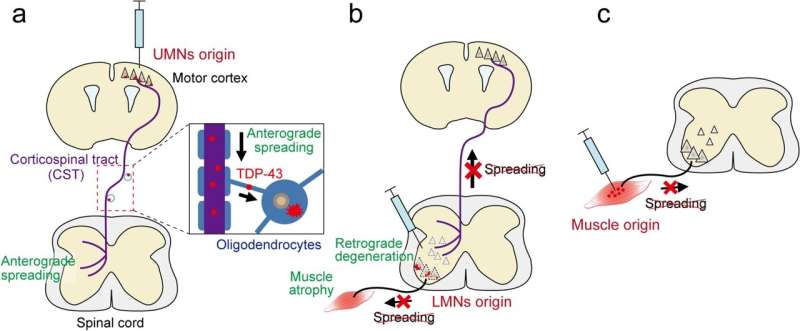 Les auteurs ont établi de nouveaux modèles de SLA de souris qui induisaient initialement des inclusions de TDP-43 mutant dans des types neuronaux ou cellulaires spécifiques dans les circuits moteurs, et ils ont étudié si le TDP-43 et les processus pathologiques pertinents se propageaient à travers les connexions neuronales ou cellulaires.
Les auteurs ont établi de nouveaux modèles de SLA de souris qui induisaient initialement des inclusions de TDP-43 mutant dans des types neuronaux ou cellulaires spécifiques dans les circuits moteurs, et ils ont étudié si le TDP-43 et les processus pathologiques pertinents se propageaient à travers les connexions neuronales ou cellulaires.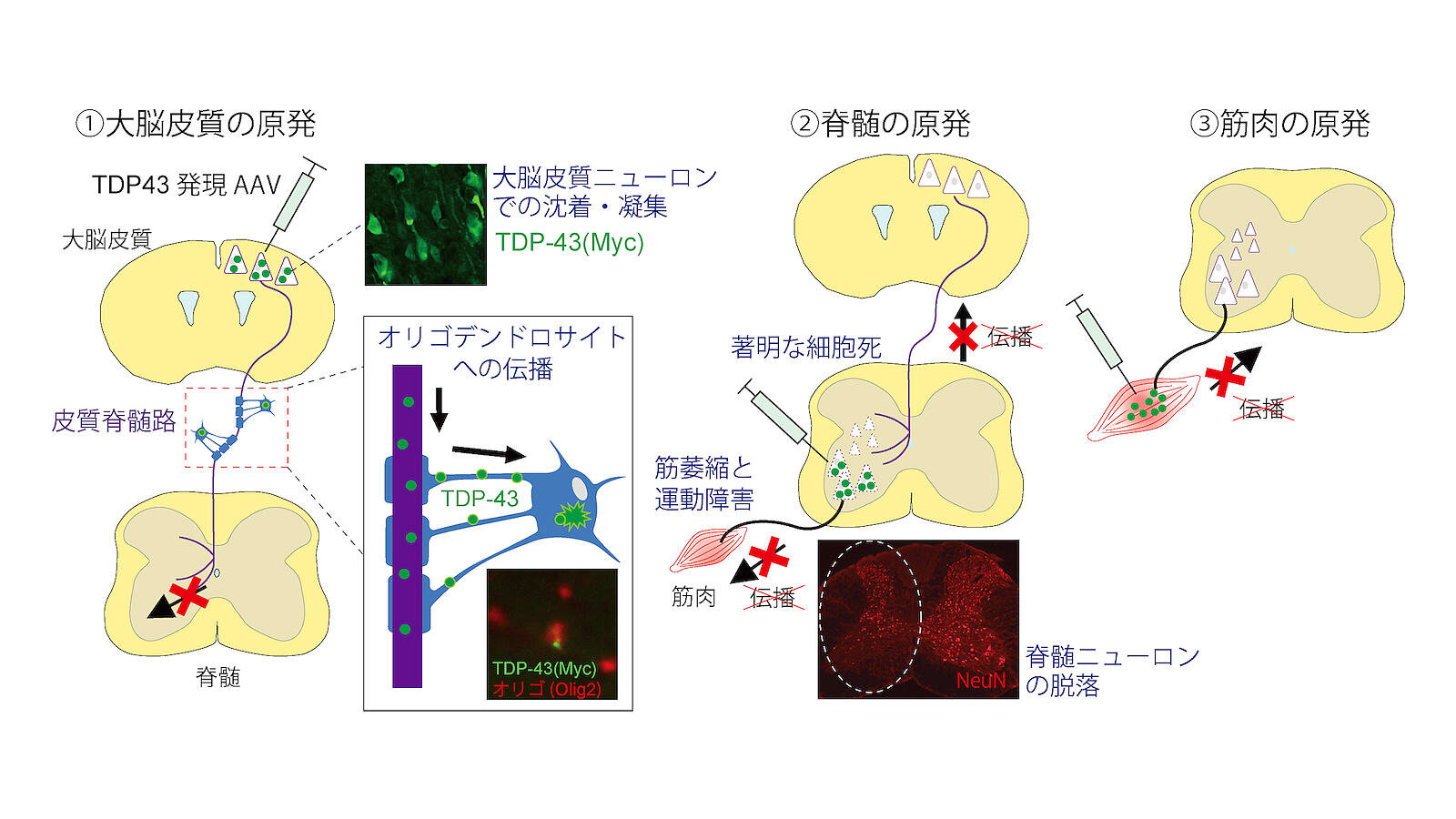 Tsuboguchi et al., Acta Neuropathologica 2023.
Tsuboguchi et al., Acta Neuropathologica 2023. Cependant, il reste difficile de savoir si cet oxystérol joue un rôle dans l'agrégation et la propagation de l'α-synucléine.
Cependant, il reste difficile de savoir si cet oxystérol joue un rôle dans l'agrégation et la propagation de l'α-synucléine. 
 La FDA et l'agence Européenne des médicaments ont exprimés de nombreux doutes sur l'efficacité de ce médicament au cours de l'essai CENTAUR.
Ces doutes concernent l'exclusion d'un certain nombre d'évènements défavorables lors de l'analyse statistique, ainsi que le fait que certains patients ont aussi reçu du Riluzole et de l'Edaravone.
En particulier il y a plus de patients ayant reçu de l'Edaravone dans la branche contrôle que dans la branche traitement, or certains scientifiques et agences du médicament pensent que l'Edaravone a un effet négatif sur l'évolution de la maladie. Certains médecins ont également attribué les "bons" résultats au TUDCA.
Au final à 24 mois il n'y a pas d'amélioration de la survie.
La FDA et l'agence Européenne des médicaments ont exprimés de nombreux doutes sur l'efficacité de ce médicament au cours de l'essai CENTAUR.
Ces doutes concernent l'exclusion d'un certain nombre d'évènements défavorables lors de l'analyse statistique, ainsi que le fait que certains patients ont aussi reçu du Riluzole et de l'Edaravone.
En particulier il y a plus de patients ayant reçu de l'Edaravone dans la branche contrôle que dans la branche traitement, or certains scientifiques et agences du médicament pensent que l'Edaravone a un effet négatif sur l'évolution de la maladie. Certains médecins ont également attribué les "bons" résultats au TUDCA.
Au final à 24 mois il n'y a pas d'amélioration de la survie.