A clinical trial (NCT04866771) was conducted at the University of Illinois Chicago to investigate the effects of remotely supervised transcranial direct current stimulation (tele-tDCS) on ALS patients. By enabling patients to undergo treatment in the comfort of their own homes under remote supervision, tele-tDCS promises to minimize travel-related barriers.
Patients were stratified into two groups based on their ALS Functional Rating Scale (ALSFS) score progression rate. The intervention group received 72 sessions of tele-tDCS, while the delayed-start group received 36 sham sessions followed by 36 active sessions. Out of 70 individuals initially screened, 14 (7 males, 7 females) were enrolled but only 10 participants completed the study. The intervention group had full retention, while the delayed-start group had a 57% retention rate.
Assessments were conducted at six-time points: pre-testing (T0), up to three mid-testing sessions (T1), post-testing at 24 weeks (T2), and a follow-up at three months (T3). These evaluations included functional and neurophysiological tests, as well as clinical and scalp integrity checks.
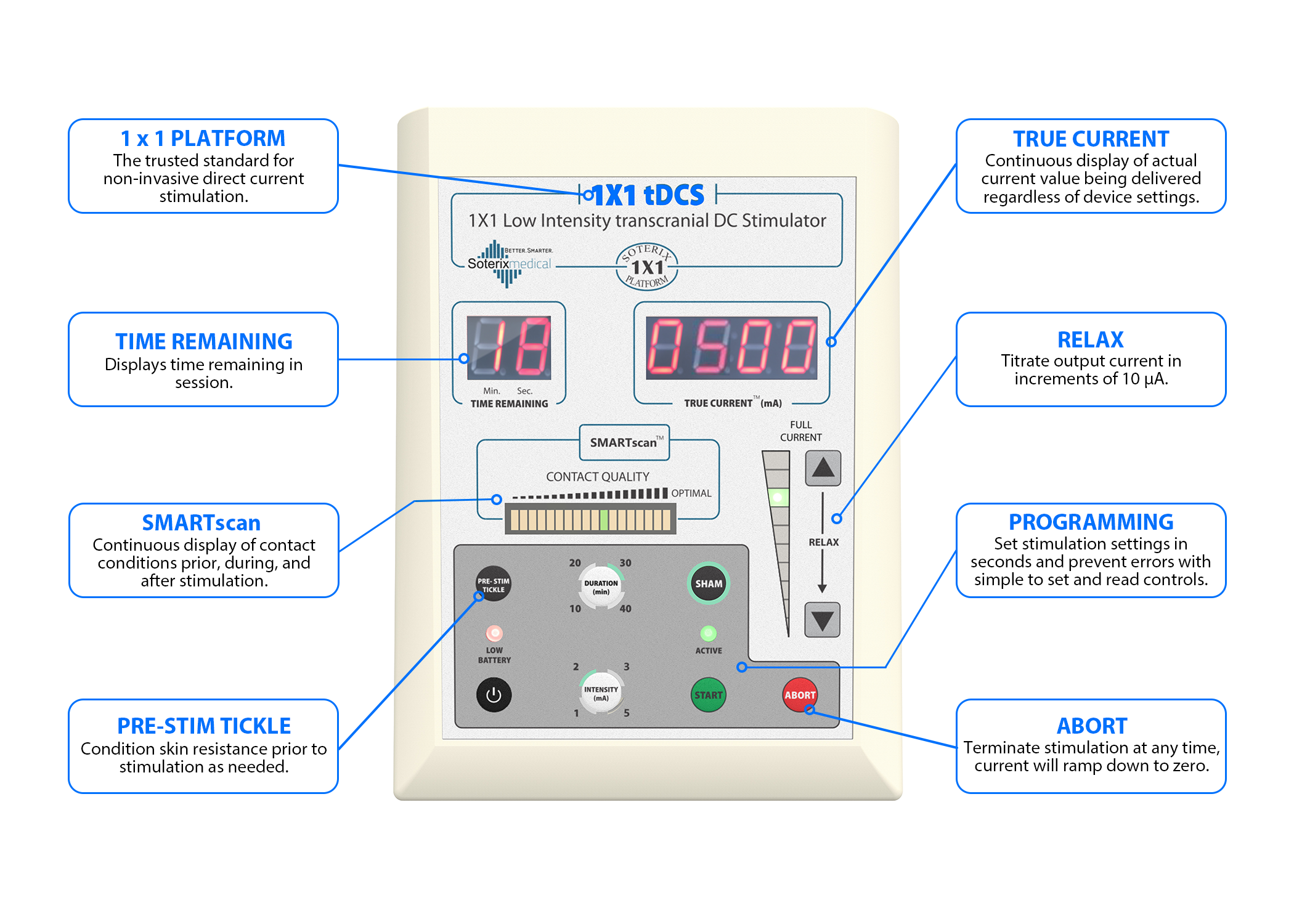
Tele-tDCS was administered three times per week for 24 weeks, with a stimulation dosage of 2 mA for 20 minutes. The devices were preprogrammed to ensure consistency and prevent alterations by participants or caregivers.
All intervention sessions were facilitated via ZoomPHI, allowing the participant and the researcher to see each other throughout the process. A caregiver was required always to be present to start and stop the session as instructed, ensuring safety and proper operation. Training was provided to ensure correct headset placement and operation, and caregivers were required to assist in starting and stopping each session.
A portable tDCS device (Soterix Medical 1X1 tDCS mini-CT Stimulator, NY) was used in this study. This device included a stimulator, a customized head strap for secure placement, and designated positions for active (anodal current over the lower limb motor cortex) and inactive electrodes (cathodal current over the contralateral supraorbital region).
It featured built-in programmable codes, allowing for controlled session-specific settings under the remote supervision of a researcher. The stimulation dosage of 2 mA for 20 min was preprogrammed into the device by research personnel before being provided to participants.
An interim analysis was conducted after six participants completed the study. The study would be halted for review if the mean ALSFS-score difference between groups exceeded two standard deviations. The "two standard deviations" rule is a way to check if the observed difference between groups is improbable. Participants were categorized as slow, intermediate, or fast progressors based on these rates.
ALSFRS-R scores at the beginning did not significantly differ between groups.
 Some people in the intervention group showed an astonishingly slower disease progression compared to the delayed-start group:
Some people in the intervention group showed an astonishingly slower disease progression compared to the delayed-start group:
From pre-testing to post-testing at 24 weeks the intervention group mean change was 1.7 (only a little degradation in ALSFR), while in the delayed-start group, there was a 13.6 change. However it looks like the situation in the intervention group was not homogeneous at all, there were patients who reacted extremely well to the therapy, while others reacted extremely badly to the therapy.
Statistically results from a group of 14 people mean absolutely nothing, yet ALS is without cure and this result is much better than in any other ALS clinical trial.
As noted by the authors future studies may benefit from incorporating objective biomarkers such as NFL to assess the effects.
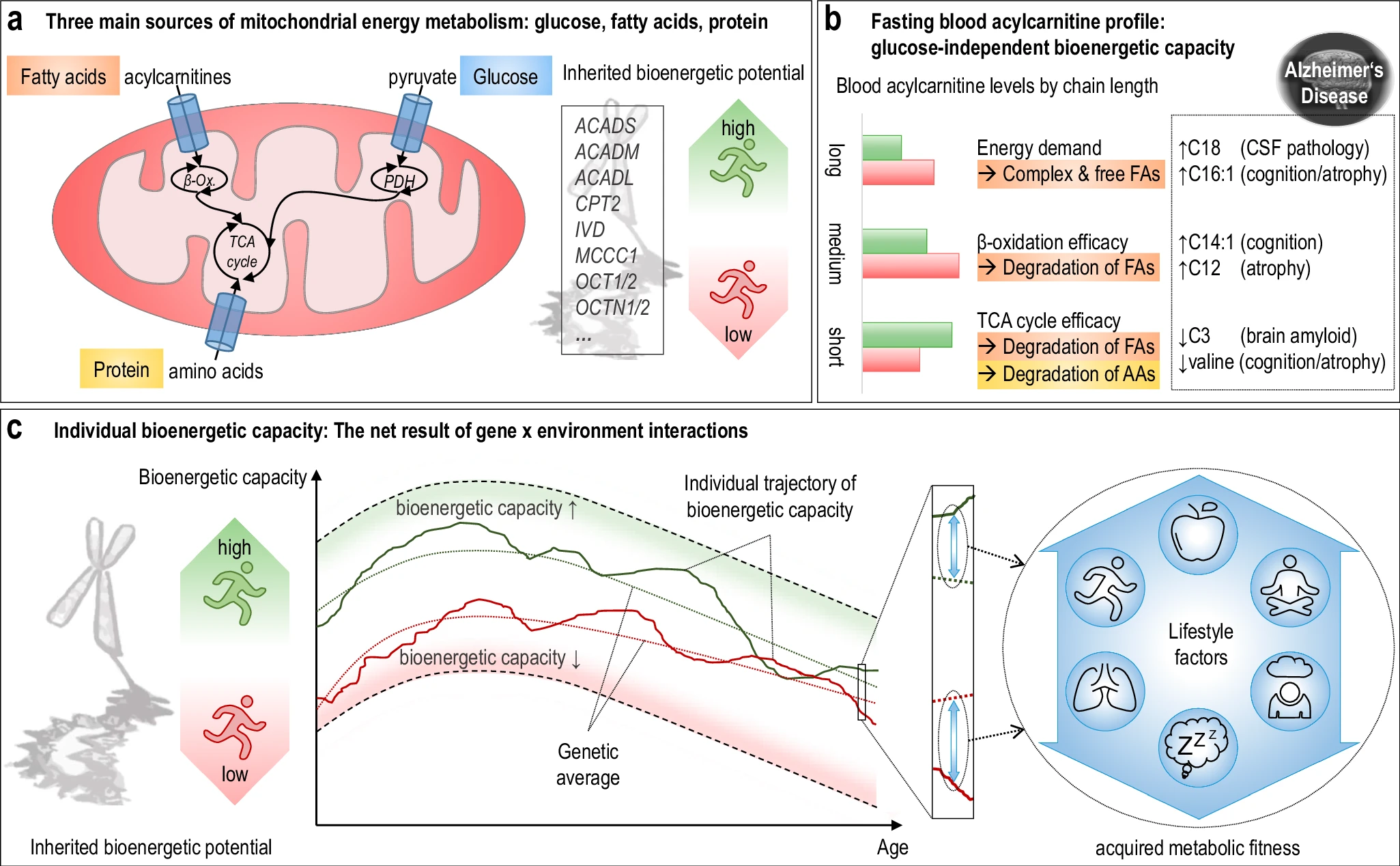 The researchers used acylcarnitine profiles from blood samples to identify distinct bioenergetic subgroups in Alzheimer's Disease (AD) patients and evaluate how bioenergetic capacity relates to disease progression.
They used data from 1,531 participants in the Alzheimer's Disease Neuroimaging Initiative (ADNI), and identified several bioenergetic subgroups with significant differences in AD biomarkers, cognitive function, and brain glucose metabolism.
These subgroups were primarily determined by modifiable factors (40-60%) related to beta-oxidation function, rather than genetic factors, suggesting potential for intervention.
The researchers used acylcarnitine profiles from blood samples to identify distinct bioenergetic subgroups in Alzheimer's Disease (AD) patients and evaluate how bioenergetic capacity relates to disease progression.
They used data from 1,531 participants in the Alzheimer's Disease Neuroimaging Initiative (ADNI), and identified several bioenergetic subgroups with significant differences in AD biomarkers, cognitive function, and brain glucose metabolism.
These subgroups were primarily determined by modifiable factors (40-60%) related to beta-oxidation function, rather than genetic factors, suggesting potential for intervention.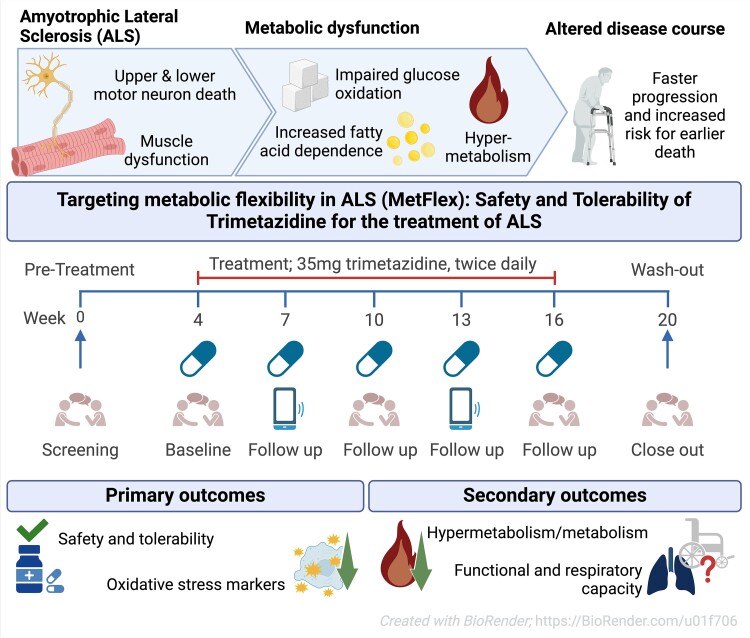 While the publication recounts that trimetazidine was beneficial for patients (this is not a phase III trial), for me the results section does not show conclusive results. For example, the results improved only during the wash-out period.
While the publication recounts that trimetazidine was beneficial for patients (this is not a phase III trial), for me the results section does not show conclusive results. For example, the results improved only during the wash-out period.