Les systèmes de navigation oculaire vous permettent de contrôler votre chaise roulante, votre domotique, un robot manipulateur et d'autres objets au moyen d'une caméra qui observe où vous regardez et transcrit vos commandes sur un écran d'ordinateur qui in-fine contrôle le fauteuil roulant ou la domotique. Il est très difficile de se procurer un tel fauteuil dans le commerce. Il existe de nombreuses publications de scientifiques à ce sujet, mais celles qui sont utilisables par une personne en situation de handicap sont en nombre infime et la plupart des études réalistes ne décrivent pas une adaptation à un fauteuil électrique du commerce.
L'un des projets les plus réalistes est celui d'une personne malade de la SLA (et décédée depuis) qui s'appelle Patrick Joyce.
- https://hackaday.io/project/5426-eye-controlled-wheelchair
- https://github.com/Sean3Don/Eyedrivomatic-github
L'adaptation du système à un fauteuil commercial se fait par une sorte de coiffe robotisée qui s'adapte par dessus le joystick du fauteuil et qui manipule celui-ci sous le contrôle d'un programme. Le programme "observe" la position des rétines de l'utilisateur ainsi que les clignements d'yeux. L'utilisateur lui, regarde un écran où différentes icones correspondent à différentes actions (aller à gauche, à droite, arrêt, etc). A partir de la position des yeux, le programme déduit quels icones sont regardées et donc quelles sont les commandes qui sont activées par l'utilisateur. Attention le principe utilisé pour commander le joystick, s'il est universel, n'est pas très précis. Le programme lui-même doit être adapté à l'ordinateur qui l'exécute. Ce n'est donc pas un système clé-en-main.
Comme d'autres méthodes de contrôle alternatives (casque EEG, détection de contraction musculaire), un système de navigation oculaire ne sera jamais aussi précis que le contrôle manuel traditionnel. Bien que les systèmes de navigation oculaire puissent être utilisé à l'extérieur, il est fortement recommandé de ne les utiliser qu'en intérieur. La principale raison est due aux limites des caméras de navigation oculaires qui fonctionnent en infra-rouge. Elles ne fonctionnent donc pas de manière fiable en plein soleil, proche d'une source de chaleur ni même dans des conditions nuageuses.
Il y a eu apparement une volonté de Patrick Joyce de diffuser largement son projet, mais son décès semble avoir stopper les efforts de la communauté qu'il avait créé via Hackaday.
Un autre projet réaliste est celui de Bob Paradiso.
https://bobparadiso.com/2015/06/16/eye-gaze-controlled-wheelchair-robotic-arm-and-ecu/
https://github.com/bobparadiso/wheelchairEyeGazeECU/tree/master
Le principe est le même que celui de Patrick Joyce mais ici le circuit électronique se raccorde sur le connecteur de raccordement du joystick au fauteuil. Bien que plus ancien que celui de Patrick Joyce, ce projet est plus évolué, outre la commande du fauteuil, il y a un bras domotique.
Bien que le protocole de Joystick soit relativement standardisé, il n'est pas certain que tous les fauteuils l'implémente. Par contre la précision est forcément meilleure que dans le projet de Patrick Joyce. Avant d'envisager d'utiliser tel ou tel fauteuil, il faut donc s'assurer que le joystick utilise un protocole et un connecteur standard. Mode digital (pas analogique) PIN 1: Avant PIN 2: Arrière PIN 3: Gauche Pin 4: Droit PIN 5: DETECT PIN 6: 5 bouton PINS 7 AND 9 – 12V 100MA PIN 8 – GROUND
Le controleur Arduino pilote directement les moteurs du bras robotique et les émetteurs IR/RF qui contrôlent les appareils environnants.
Une amélioration évidente de ce projet serait de piloter le fauteuil roulant via Bluetooth. En effet, certains fauteuils roulants modernes permettent un contrôle à distance via Bluetooth, il peut donc être possible d'interfacer le projet de Bob Paradiso à de tels fauteuils roulants sans avoir à interférer avec leur électronique.
D'autres projets intéressants sont:
 In the case of ALS, the clusters of TDP-43 are also located in the cytosol, i.e. where the proteins are produced, before being folded up in the ER and then sent to their place of use by the Golgi apparatus. After shipment, TDP-43 should be in the nucleus.
In the case of ALS, the clusters of TDP-43 are also located in the cytosol, i.e. where the proteins are produced, before being folded up in the ER and then sent to their place of use by the Golgi apparatus. After shipment, TDP-43 should be in the nucleus. Des preuves cliniques et épidémiologiques établissent un lien entre les altérations métaboliques et l'apparition et la progression de la sclérose latérale amyotrophique. Ces défauts métaboliques précèdent les symptômes moteurs, ce qui suggére que ces défauts sont au moins en partie à l'origine de la SLA.
Des preuves cliniques et épidémiologiques établissent un lien entre les altérations métaboliques et l'apparition et la progression de la sclérose latérale amyotrophique. Ces défauts métaboliques précèdent les symptômes moteurs, ce qui suggére que ces défauts sont au moins en partie à l'origine de la SLA. Des scientifiques se sont demandé quel était l'impact de l'exposition aux stéroïdes endogènes et synthétiques chez les patientes atteinte de la SLA. Ils ont comparé cet impact à celui d'un groupe comparable mais ne souffrant pas de la SLA.
Des scientifiques se sont demandé quel était l'impact de l'exposition aux stéroïdes endogènes et synthétiques chez les patientes atteinte de la SLA. Ils ont comparé cet impact à celui d'un groupe comparable mais ne souffrant pas de la SLA.
 The authors initially hypothesized that activation of the inhibitory co-receptor PD-1 by recombinant PD-L1 would be anti-inflammatory. However, recombinant PD-L1 ligand and recombinant PD-1 receptor were strongly pro-inflammatory.
Then they abandoned the PD-L1 strategy and chose a strategy with dimethyl fumarate (DMF), a drug approved against two autoimmune diseases, multiple sclerosis and psoriasis, and the cGAS- STING H-151 involved in autoimmunity.
The authors initially hypothesized that activation of the inhibitory co-receptor PD-1 by recombinant PD-L1 would be anti-inflammatory. However, recombinant PD-L1 ligand and recombinant PD-1 receptor were strongly pro-inflammatory.
Then they abandoned the PD-L1 strategy and chose a strategy with dimethyl fumarate (DMF), a drug approved against two autoimmune diseases, multiple sclerosis and psoriasis, and the cGAS- STING H-151 involved in autoimmunity. Les culturistes utilisent régulièrement le β-hydroxy β-méthylbutyrate (HMB) ou le produit voisin, l'acide β-hydroxy β-méthylbutyrique (HMB-FA) comme supplément de renforcement musculaire pour augmenter les gains de taille et de force musculaire induits par l'exercice et améliorer les performances physiques.
Les culturistes utilisent régulièrement le β-hydroxy β-méthylbutyrate (HMB) ou le produit voisin, l'acide β-hydroxy β-méthylbutyrique (HMB-FA) comme supplément de renforcement musculaire pour augmenter les gains de taille et de force musculaire induits par l'exercice et améliorer les performances physiques.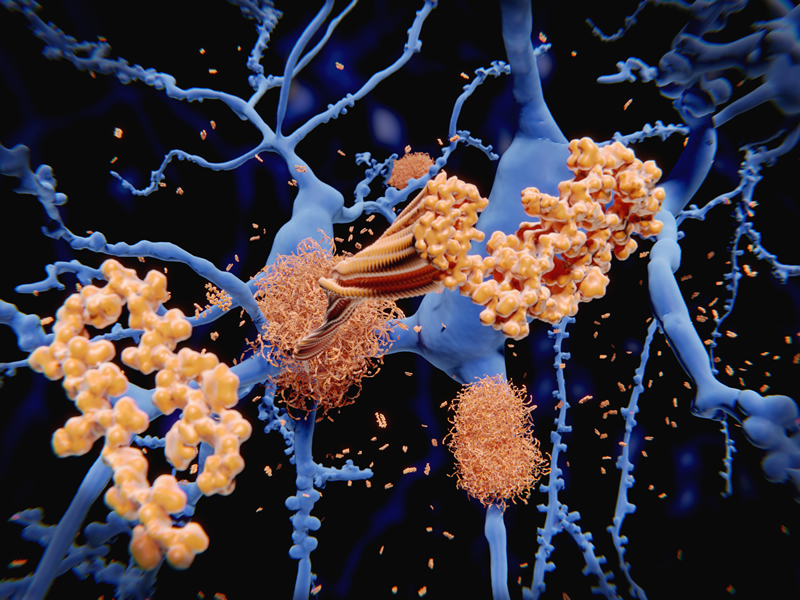 Naphthoquinones are antiprotozoal drugs. Protozoa are small organisms, which exist as solitary cells or colonies of cells. They live in water or in moist soil or inside an organism (in the lung mucus, the intestine, the paunch of certain animals, etc.). They are known to be responsible for many diseases such as malaria and certain dysentery, such as amoebosis.
Naphthoquinones are antiprotozoal drugs. Protozoa are small organisms, which exist as solitary cells or colonies of cells. They live in water or in moist soil or inside an organism (in the lung mucus, the intestine, the paunch of certain animals, etc.). They are known to be responsible for many diseases such as malaria and certain dysentery, such as amoebosis.