Nouvelle perspective sur la SLA : Comment les muscles réguleraient la protéine TDP-43 au niveau de la synapse.
Pendant de nombreuses années, la SLA liée à la SOD1 a été considérée comme un cas particulier au sein du spectre de la maladie : un sous-type de SLA où la protéine TDP-43, qui caractérise la pathologie de la plupart des cas de SLA, jouerait un rôle mineur, voire inexistant. Cette hypothèse est aujourd’hui remise en question. Une étude récente révèle que la protéine TDP-43 est fortement impliquée dans la SLA liée à la SOD1, mais d’une manière qui avait échappé aux observations précédentes : la pathologie se situerait non pas dans les corps cellulaires des motoneurones, mais dans les axones et à la jonction neuromusculaire (JNM).
Ce changement de perspective modifie notre compréhension de la pathologie précoce de la SLA et ouvre de nouvelles pistes thérapeutiques axées sur la synapse, voire sur le muscle lui-même.
 Lorsque les pathologistes examinent les motoneurones spinaux de patients atteints de SLA liée à SOD1, les noyaux apparaissent généralement normaux : la protéine TDP-43 est toujours présente et les agrégats anormaux sont rarement observés. C’est pourquoi la SLA liée à SOD1 a été considérée comme « TDP-43 négative ».
Lorsque les pathologistes examinent les motoneurones spinaux de patients atteints de SLA liée à SOD1, les noyaux apparaissent généralement normaux : la protéine TDP-43 est toujours présente et les agrégats anormaux sont rarement observés. C’est pourquoi la SLA liée à SOD1 a été considérée comme « TDP-43 négative ».
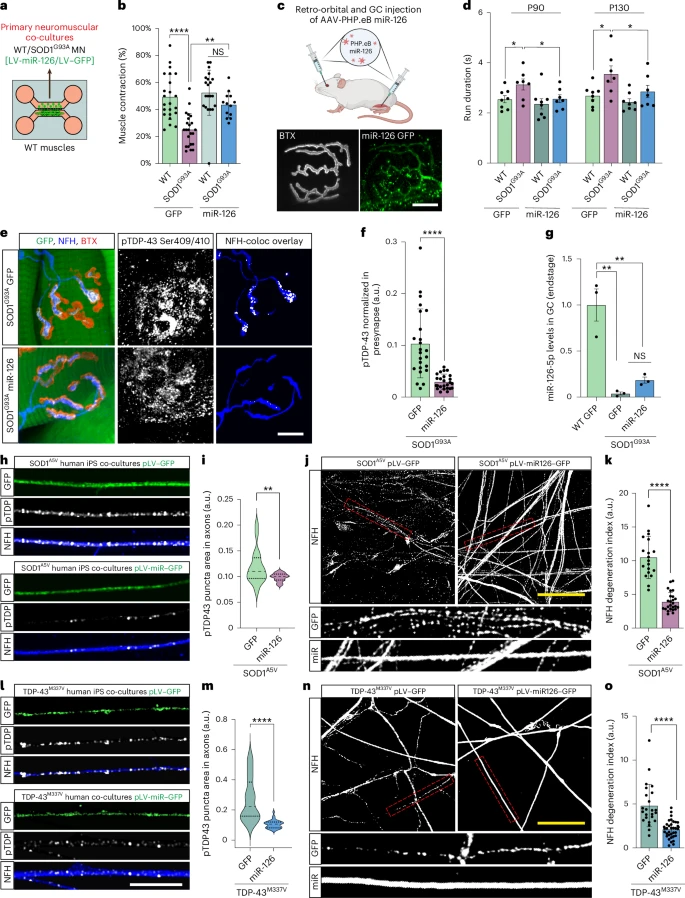
Cependant, cette étude révèle que la situation est très différente en périphérie. Chez les patients atteints de SLA liée à SOD1 et dans les modèles murins SOD1 (G93A et G37R) : La protéine TDP-43 phosphorylée forme des agrégats dans les axones moteurs périphériques. La protéine TDP-43 s’accumule précocement à la jonction neuromusculaire, bien avant l’apparition des symptômes. Les corps cellulaires des motoneurones restent normaux, avec une protéine TDP-43 nucléaire intacte.
Ce schéma représente une forme non canonique de pathologie TDP-43. Les lésions sont concentrées au niveau des synapses au bout du très long axone, et non du soma. Cette distinction est importante car la SLA débute souvent par une dégénérescence régressive, la détérioration de la jonction neuromusculaire (JNM) précédant la mort des motoneurones. Les motoneurones dépendent fortement de la traduction locale pour le maintien des mitochondries, des vésicules et des éléments du cytosquelette. Un excès de TDP-43 à la terminaison inhibe ces processus.
Synthèse locale de TDP-43 : une vulnérabilité insoupçonnée
Les axones moteurs sont extrêmement longs et dépendent de la synthèse protéique locale pour maintenir leurs terminaisons. L’étude ci-dessus montre que la TDP-43 elle-même est une de ces protéines régulées localement. Dans des conditions normales, cette traduction est maintenue à un faible niveau.
Ce pool local de TDP-43 semble inoffensif lorsqu’il est étroitement contrôlé. Mais lorsque sa régulation est perturbée, l’axone devient vulnérable à un excès de TDP-43 et à sa capacité connue à inhiber la traduction de nombreux autres ARNm.
L'étude montre que muscle inhibe activement la TDP-43 présynaptique via les exosomes. Il n'est pas comme on le croyait jusque là, un acteur passif dans la biologie de la JNM. Le muscle libère des vésicules extracellulaires (VE) chargées de molécules régulatrices qui influencent l'axone moteur. Ces VE d'origine musculaire contiennent miR-126-5p, un microARN qui réprime fortement la traduction de TDP-43, AGO2 et d'autres composants des voies de silençage de l'ARN.
Les axones moteurs de la jonction neuromusculaire (JNM) absorbent ces vésicules, ce qui contribue à contrôler la synthèse locale de TDP-43. Le muscle exerce ainsi une influence trans-synaptique protectrice sur le neurone.
Dans la SLA, ce système protecteur est défaillant.
Dans la SLA liée à SOD1, l'étude révèle une forte diminution du taux de miR-126-5p, lorsque le taux de miR-126-5p chute, l'inhibition de la production locale de TDP-43 est levée. Il y a alors une synthèse excessive de TDP-43 au niveau des terminaisons axonales présynaptiques, une diminution de la synthèse protéique locale et à terme une défaillance et dégénérescence de la jonction neuromusculaire (JNM). Les motoneurones sont structurellement fragiles. La longueur considérable de ces cellules les rend particulièrement dépendantes de la synthèse protéique locale. Un blocage localisé de cette synthèse peut entraîner une dénervation, même si le soma est intact.
Ce mécanisme établit un lien direct entre la dégénérescence précoce de la JNM et la toxicité du TDP-43, même lorsque ce dernier n’a pas encore quitté le noyau.
Inhibition de miR-126-5p au niveau de la jonction neuromusculaire
La toxicité de TDP-43 est généralement associée à sa perte nucléaire : TDP-43 quitte le noyau, ses fonctions normales de maturation de l'ARN sont altérées, et des lésions de l'ADN ou des erreurs d'épissage s'ensuivent. Cette étude met en lumière un autre problème : un excès local de TDP-43 peut être nocif même lorsque le TDP-43 nucléaire est intact.
Les auteurs ont testé leur hypothèse en bloquant la libération de vésicules extracellulaires (VE) par le muscle. Ces manipulations ont produit les mêmes effets que ceux observés dans la SLA : augmentation du TDP-43 axonal, réduction de la traduction locale et dégénérescence de la JNM. Fait important, l’administration d’ARNsi ciblant le TDP-43 a empêché cette dégénérescence, démontrant que la surabondance de TDP-43 au niveau de la synapse en est le facteur déterminant.
Stimuler le miR-126 peut améliorer la fonction de la jonction neuromusculaire (JNM)
Lorsque les chercheurs ont rétabli les niveaux de miR-126 chez des souris SOD1, la structure et la fonction de la JNM se sont améliorées et les marqueurs pathologiques ont diminué. Bien qu'il s'agisse d'expériences thérapeutiques préliminaires, elles ouvrent la voie à de nouvelles stratégies d'intervention agissant au niveau de la synapse plutôt qu'au niveau du noyau.
Implications thérapeutiques
L'étude suggère plusieurs pistes de traitement, chacune nécessitant une évaluation rigoureuse et réaliste, mai surtout le muscle plutôt que le neurone moteur devient une cible thérapeutique.
Traditionnellement, les traitements de la SLA ciblent le neurone. Or, le muscle apparaît comme un site d'intervention prometteur, car il régule naturellement l'homéostasie des protéines présynaptiques via les vésicules extracellulaires (VE). Étant donné que le blocage de la sécrétion des VE accélère la dégénérescence, le maintien d'un trafic sain de VE pourrait avoir des effets protecteurs. Si l'accumulation synaptique est un événement précoce et distinctif, les interventions visant à éliminer ou à réduire ces agrégats – y compris les stratégies antisens – pourraient s'avérer précieuses, même dans les types de SLA que l'on pensait auparavant indépendants de la protéine TDP-43.
Convergence entre les sous-types de SLA
Plus largement la découverte de la pathologie TDP-43 dans la SLA liée à SOD1 suggère des mécanismes en aval communs à plusieurs variantes de la SLA. Ceci pourrait permettre d'unifier les approches thérapeutiques plutôt que de les fragmenter selon le génotype.
Cette étude renouvelle notre compréhension de la protéine TDP-43 dans la SLA, en particulier dans les modèles SOD1. Au lieu d'un problème nucléaire au niveau du motoneurone, les lésions résultent d'une dérégulation de la traduction locale au niveau de la synapse. Le rôle habituel du muscle dans la limitation de la production de TDP-43 est altéré, ce qui permet la formation d'agrégats toxiques à la jonction neuromusculaire et affaiblit la connexion entre le nerf et le muscle.
En mettant en lumière un mécanisme précoce, agissant en dehors du système nerveux central, ces travaux ouvrent la voie à des stratégies thérapeutiques à la fois novatrices et potentiellement plus accessibles : la restauration du miR-126 d'origine musculaire, le soutien de la signalisation par vésicules extracellulaires et le ciblage du TDP-43 synaptique avant qu'il ne déstabilise l'ensemble de l'unité motrice.
Si de futures études confirment ces résultats, la jonction neuromusculaire – voire le muscle lui-même – pourrait constituer l'une des cibles les plus prometteuses pour une intervention précoce dans la SLA.