New Perspective on ALS: How Muscles Regulate TDP-43 at the Synapse
For many years, SOD1-related ALS was considered a special case within the disease spectrum: a subtype of ALS where the TDP-43 protein, which characterizes the pathology of most ALS cases, played a minor or even nonexistent role. This hypothesis is now being challenged. A recent study reveals that the TDP-43 protein is strongly implicated in SOD1-related ALS, but in a way that had escaped previous observations: the pathology appears to be located not in the cell bodies of motor neurons, but in the axons and at the neuromuscular junction (NMJ).
This shift in perspective alters our understanding of the early pathology of ALS and opens new therapeutic avenues focused on the synapse, or even on the muscle itself.
 When pathologists examine the spinal motor neurons of patients with SOD1-related ALS, the nuclei generally appear normal: the TDP-43 protein is always present, and abnormal aggregates are rarely observed. This is why SOD1-related ALS has been considered "TDP-43 negative."
When pathologists examine the spinal motor neurons of patients with SOD1-related ALS, the nuclei generally appear normal: the TDP-43 protein is always present, and abnormal aggregates are rarely observed. This is why SOD1-related ALS has been considered "TDP-43 negative."
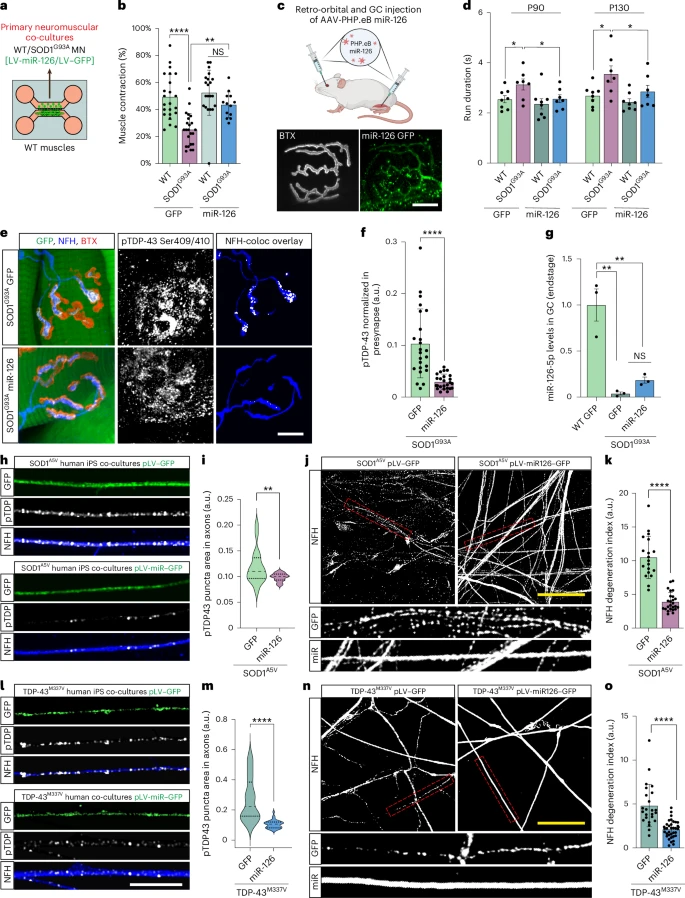
However, this study reveals that the situation is very different in the periphery. In patients with SOD1-related ALS and in SOD1 mouse models (G93A and G37R): Phosphorylated TDP-43 protein forms aggregates in peripheral motor axons. TDP-43 protein accumulates early at the neuromuscular junction, well before the onset of symptoms. Yet the cell bodies of the motor neurons remain normal, with intact nuclear TDP-43 protein.
This article represents a non-canonical form of TDP-43 pathology. The lesions are concentrated at the synapses at the ends of the very long axons, not at the soma. This distinction is important because ALS often begins with regressive degeneration, with deterioration of the neuromuscular junction (NMJ) preceding motor neuron death. Motor neurons rely heavily on local translation for the maintenance of mitochondria, vesicles, and cytoskeletal components. An excess of TDP-43 at the terminal inhibits these processes.
Local TDP-43 Synthesis: An Unsuspected Vulnerability
Motor axons are extremely long and depend on local protein synthesis to maintain their terminals. The study above shows that TDP-43 itself is one of these locally regulated proteins. Under normal conditions, this translation is maintained at a low level.
This local pool of TDP-43 appears harmless when tightly controlled. But when its regulation is disrupted, the axon becomes vulnerable to an excess of TDP-43 and its known ability to inhibit the translation of many other mRNAs.
The study shows that muscle actively inhibits presynaptic TDP-43 via exosomes. The muscle is not, as previously thought, a passive player in the biology of the neuromuscular junction (NMJ). Muscle releases extracellular vesicles (EVs) loaded with regulatory molecules that influence the motor axon. These muscle-derived EVs contain miR-126-5p, a microRNA that strongly represses the translation of TDP-43, AGO2, and other components of RNA silencing pathways.
Motor axons at the neuromuscular junction (NMJ) take up these vesicles, which helps control local TDP-43 synthesis. Muscle thus exerts a protective trans-synaptic influence on the neuron.
In ALS, this protective system malfunctions.
In SOD1-related ALS, the study reveals a sharp decrease in miR-126-5p levels. When miR-126-5p levels drop, the inhibition of local TDP-43 production is lifted. This leads to excessive TDP-43 synthesis at presynaptic axonal terminals, decreased local protein synthesis, and ultimately, failure and degeneration of the neuromuscular junction (NMJ). Motor neurons are structurally fragile. Their considerable length makes them particularly dependent on local protein synthesis. Localized blockage of this synthesis can lead to denervation, even if the soma is intact.
This mechanism establishes a direct link between early NMJ degeneration and TDP-43 toxicity, even when the latter has not yet left the nucleus.
Inhibition of miR-126-5p at the neuromuscular junction
TDP-43 toxicity is generally associated with its nuclear loss: TDP-43 leaves the nucleus, its normal RNA maturation functions are impaired, and DNA damage or splicing errors ensue. This study highlights another problem: a local excess of TDP-43 can be harmful even when nuclear TDP-43 is intact.
The authors tested their hypothesis by blocking the release of extracellular vesicles (EVs) are released from the muscle. These manipulations produced the same effects as those observed in ALS: increased axonal TDP-43, reduced local translation, and neuromuscular junction (NMJ) degeneration. Importantly, administration of siRNAs targeting TDP-43 prevented this degeneration, demonstrating that TDP-43 overabundance at the synapse is the determining factor.
Stimulating miR-126 can improve neuromuscular junction (NMJ) function
When researchers restored miR-126 levels in SOD1 mice, NMJ structure and function improved, and pathological markers decreased. Although these are preliminary therapeutic experiments, they pave the way for new intervention strategies that act at the synapse rather than the nucleus.
Therapeutic Implications
The study suggests several treatment avenues, each requiring rigorous and realistic evaluation, but above all, muscle rather than the motor neuron is emerging as a therapeutic target.
Traditionally, ALS treatments have targeted the neuron. However, muscle appears as a promising site of intervention because it naturally regulates presynaptic protein homeostasis via extracellular vesicles (EVs). Given that blocking EV secretion accelerates degeneration, maintaining healthy EV trafficking could have protective effects. If synaptic accumulation is an early and distinctive event, interventions aimed at eliminating or reducing these aggregates—including antisense strategies—could prove valuable, even in ALS types previously thought to be independent of the TDP-43 protein.
Convergence Among ALS Subtypes
More broadly, the discovery of TDP-43 pathology in SOD1-related ALS suggests downstream mechanisms common to several ALS variants. This could allow for the unification of therapeutic approaches rather than their fragmentation based on genotype.
This study renews our understanding of the TDP-43 protein in ALS, particularly in SOD1 models. Instead of a nuclear problem at the motor neuron level, the lesions result from a dysregulation of local translation at the synapse. The usual role of muscle in limiting TDP-43 production is altered, allowing the formation of toxic aggregates at the neuromuscular junction and weakening the connection between the nerve and muscle.
By highlighting an early mechanism acting outside the central nervous system, this work paves the way for both innovative and potentially more accessible therapeutic strategies: restoring the muscle-derived miR-126, supporting signaling via extracellular vesicles, and targeting synaptic TDP-43 before it destabilizes the entire motor unit.
If future studies confirm these results, the neuromuscular junction—or even the muscle itself—could represent one of the most promising targets for early intervention in ALS.
 To understand this major breakthrough, it's helpful to dispel a common misconception about DNA damage. Our genetic code isn't a static library; it constantly undergoes routine damage. Every time a brain cell processes oxygen, generates metabolic energy, or emits an electrical impulse, it creates oxidative stress that naturally breaks its own DNA strands.
To understand this major breakthrough, it's helpful to dispel a common misconception about DNA damage. Our genetic code isn't a static library; it constantly undergoes routine damage. Every time a brain cell processes oxygen, generates metabolic energy, or emits an electrical impulse, it creates oxidative stress that naturally breaks its own DNA strands.
 By testing this approach on mouse models of both diseases and on human patient stem cells (iPSCs), the team observed several crucial improvements:
By testing this approach on mouse models of both diseases and on human patient stem cells (iPSCs), the team observed several crucial improvements: