There are often many causes to a non-communicable disease, particularly neurodegenerative diseases are more a consequence of a systemic failure than caused by a specific phenomenon. The multitude of papers assigning a specific mechanism, each time different, to neurodegenerative diseases is just noise that drags down knowledge acquisition in these domains. Some authors have hinted at a phase transition to explain the misfolding of some proteins, but what triggers this phase transition was elusive.
In this post, I discuss a very general paper.
https://elifesciences.org/reviewed-preprints/107962v1
In simple terms, the authors have discovered how our innate immune system launches an extremely powerful and rapid response to a tiny signal from a pathogen. This has implications for age-related diseases such as cancer or neurodegenerative diseases.
The Core Problem:
Our immune system needs to react decisively to a single bacterium or virus. This involves a massive cellular response like inflammation or programmed cell death (pyroptosis, apoptosis). However, the initial detection of a pathogen (a single molecule binding to a receptor) provides almost no energy to power this massive response.
The Discovery - "Metastable Supersaturation":
 The authors found that key immune signaling proteins, specifically those containing Death Fold Domains (DFDs) (like ASC, FADD, BCL10, MAVS, TRADD), exist in a unique physical state inside our cells called metastable supersaturation. These full-length adaptors retain nucleation barriers and are able to exist supersaturated in cells. In contrast, many receptors and effectors do not. This localizes the “spring-loaded” behaviour to central adaptors that link receptor sensing to downstream cell-fate decisions.
The authors found that key immune signaling proteins, specifically those containing Death Fold Domains (DFDs) (like ASC, FADD, BCL10, MAVS, TRADD), exist in a unique physical state inside our cells called metastable supersaturation. These full-length adaptors retain nucleation barriers and are able to exist supersaturated in cells. In contrast, many receptors and effectors do not. This localizes the “spring-loaded” behaviour to central adaptors that link receptor sensing to downstream cell-fate decisions.
A subset of death-fold domains (DFDs) are intrinsically “supersaturable.” Using a systematic screen of 109 human DFDs with a distributed amphifluoric FRET (DAmFRET) assay in yeast, the authors show that a minority of DFDs switch from soluble → assembled in a discontinuous (nucleation-limited) manner — the hallmark of a large intrinsic nucleation barrier. These discontinuous DFDs can therefore exist metastably above their saturation concentration (Csat) while remaining soluble (i.e. supersaturated).
Imagine a supersaturated solution of sugar water. It holds far more dissolved sugar than it should be able to. It remains liquid until you drop in a single sugar crystal, which instantly triggers the entire solution to crystallize.
Similarly, these DFD proteins are present in concentrations far higher than their natural solubility limit. They are kept in a soluble, "primed" state only by a high energy barrier that prevents them from spontaneously assembling (like the sugar needing a seed crystal).
This state acts as a long-term energy reservoir. The cell expends energy to produce and maintain these high levels of protein, storing potential energy for a future immune response. The authors show that tissues/cell types with shorter lifespans (e.g., monocytes) tend to express higher adaptor supersaturation than long-lived cells (neurons), suggesting a trade-off between rapid innate responsiveness and longevity. They also find conservation of nucleation barriers in distant taxa (fish, sponges, bacteria), indicating the mechanism is ancient.
How It Works for Immunity:
When a pathogen is detected (the initial signal), the pathogen-bound receptor acts as the "seed crystal." This seed triggers the instantaneous, explosive polymerization of the supersaturated adaptor proteins (like ASC or FADD). This amplification process consumes the stored energy from supersaturation, converting it into a massive biochemical signal that leads to inflammation or cell death.
This allows for a response that is immediate, decisive, and independent of the cell's current metabolic energy (which is often hijacked by pathogens).
The Trade-Off is Immunity vs. Longevity:
This mechanism comes with a cost. Maintaining a supersaturated, "primed" state means there's always a risk of a spontaneous, accidental activation (a stochastic nucleation event). This would lead to unwanted inflammation or cell death without any infection. The authors found evidence that this trade-off is real: short-lived immune cells (like monocytes) have much higher levels of supersaturation than long-lived cells (like neurons). This suggests a fundamental thermodynamic drive where the need for strong immunity may inherently limit a cell's lifespan.
The authors also showed this system is highly specific (DFDs from one pathway don't accidentally trigger others) and that the mechanism is evolutionarily ancient, found in everything from humans to sponges to bacteria, indicating its fundamental importance.
This groundbreaking discovery opens up entirely new avenues for treating a wide range of diseases by targeting this "supersaturation engine."
Autoinflammatory and Autoimmune Diseases
Examples: Crohn's disease, rheumatoid arthritis, lupus, CAPS (Cryopyrin-Associated Periodic Syndromes), type 1 diabetes.
Infectious Diseases
Examples: Sepsis, severe viral infections (e.g., COVID-19, flu).
Cancer
Application: Some cancers evade the immune system by preventing immune cells from initiating cell death (apoptosis) in cancerous cells. They might do this by interfering with the supersaturation or nucleation of proteins like FADD.
Neurodegenerative Diseases
Examples: Alzheimer's, Parkinson's, ALS.
Therapeutic Strategy: This research provides a deeper biophysical understanding of how proteins form aggregates. Insights into controlling nucleation barriers could lead to strategies for preventing the initial "seed" event that sparks the catastrophic aggregation of proteins like amyloid-beta or alpha-synuclein.
Risks, trade-offs, and practical challenges
Immunity vs longevity trade-off. The authors argue a thermodynamic tradeoff: lowering supersaturation protects cells from spontaneous death but reduces rapid responsiveness to pathogens. Therapies that blunt supersaturation may increase infection susceptibility.
Off-target/cross-seeding risk. Although the interactome is relatively specific, some cross-nucleation exists (e.g., PYD↔DED). Inhibiting one adaptor could have unintended effects on other pathways, or conversely, seeding one adaptor therapeutically could accidentally trigger another.
Drugging interfaces is hard. Filamentizing interfaces and nucleation kinetics are complex to target with small molecules; biologics or degradation approaches may be more tractable but have delivery challenges.
Temporal and quantitative control required. Because the system is switch-like, small quantitative changes in concentration or barrier height can produce large outcome differences; therapies need tight control to avoid tipping the balance toward immunodeficiency or hyperinflammation.
In conclusion
This study moves beyond simply listing the components of immune pathways to explaining the fundamental physics and energy dynamics that make them work. By understanding that immunity is powered by a "loaded spring" mechanism of metastable supersaturation, we can now think about designing much smarter, more precise drugs that either stabilize this spring (for autoimmune diseases) or trigger it on command (for cancer).

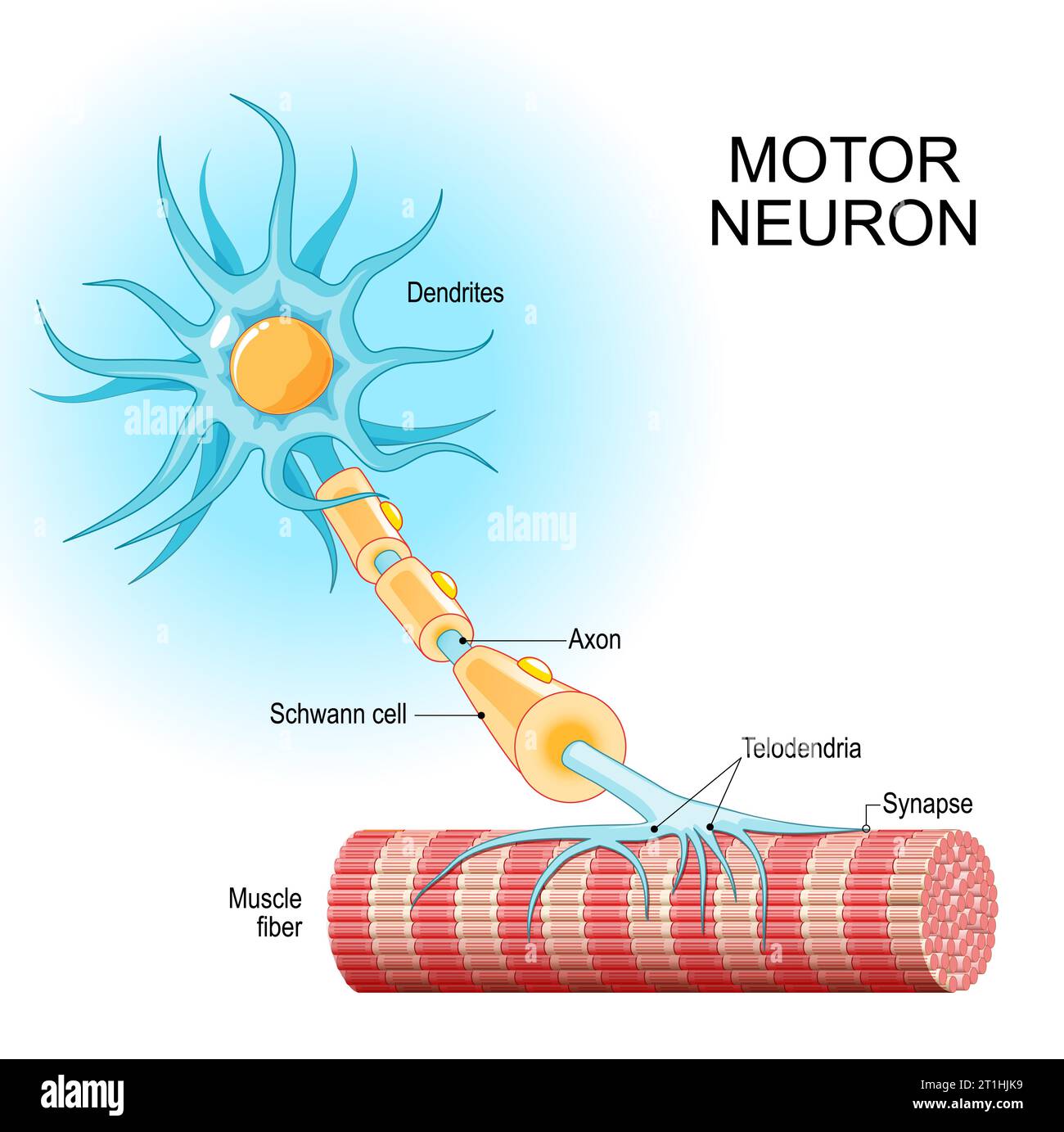
 At this stage, neuroblasts express key transcription factors like ISL1 and LHX3, which establish the fundamental identity of the motor neuron. The neuroblast begins to resemble more to a motor neuron: They extend a long axon out of the spinal cord towards their target muscle. The cell also starts to acquire its specific electrical properties. Then the neuron reaches its target muscle, forms a neuromuscular junction, and becomes a fully functional, electrically active cell. At this point, the early master regulators like ISL1 and LHX3 are largely downregulated, and the neuron enters its final, mature state.
At this stage, neuroblasts express key transcription factors like ISL1 and LHX3, which establish the fundamental identity of the motor neuron. The neuroblast begins to resemble more to a motor neuron: They extend a long axon out of the spinal cord towards their target muscle. The cell also starts to acquire its specific electrical properties. Then the neuron reaches its target muscle, forms a neuromuscular junction, and becomes a fully functional, electrically active cell. At this point, the early master regulators like ISL1 and LHX3 are largely downregulated, and the neuron enters its final, mature state.
 The authors designed a genetic therapy with an AAV virus vector to make mature neurons express two proteins that are only expressed in the immature state.
The AAVs were specifically engineered to target motor neurons. In the study conducted on mice, the administration mode of the AAV viral vector was able to specifically infect the spinal motor neurons.
Once inside the mature motor neurons, the AAV released the therapeutic genes. This caused the neurons to begin expressing ISL1 and LHX3 again
By re-expressing ISL1 and LHX3, the researchers essentially re-activate that original "immature" genetic program. This causes the mature neuron to revert to a state that is genetically and functionally similar to its younger self, with renewed resilience and stress-coping abilities.
They believe that turning on the immature genetic program essentially re-awakens the neuron's dormant ability to regrow and repair itself. While mature neurons in the central nervous system have very limited regenerative capacity, the authors are suggesting that ISL1 and LHX3 could be flipping a switch that bypasses this limitation.
The authors designed a genetic therapy with an AAV virus vector to make mature neurons express two proteins that are only expressed in the immature state.
The AAVs were specifically engineered to target motor neurons. In the study conducted on mice, the administration mode of the AAV viral vector was able to specifically infect the spinal motor neurons.
Once inside the mature motor neurons, the AAV released the therapeutic genes. This caused the neurons to begin expressing ISL1 and LHX3 again
By re-expressing ISL1 and LHX3, the researchers essentially re-activate that original "immature" genetic program. This causes the mature neuron to revert to a state that is genetically and functionally similar to its younger self, with renewed resilience and stress-coping abilities.
They believe that turning on the immature genetic program essentially re-awakens the neuron's dormant ability to regrow and repair itself. While mature neurons in the central nervous system have very limited regenerative capacity, the authors are suggesting that ISL1 and LHX3 could be flipping a switch that bypasses this limitation. De façon similaire, la force des souris modifiés génétiquement est nettement plus basse au début du traitement qu’à la fin, c’est l’inverse de ce qu’on pourrait attendre d’une souris qui serait de plus en plus affaiblie.
Et pourquoi ce groupe de souris aurait-il une force plus faible au début de l’expérience ? S’il n’y a pas de sélection à priori, les différents groupes de souris (traités et non traités) devraient avoir la même force.
De façon similaire, la force des souris modifiés génétiquement est nettement plus basse au début du traitement qu’à la fin, c’est l’inverse de ce qu’on pourrait attendre d’une souris qui serait de plus en plus affaiblie.
Et pourquoi ce groupe de souris aurait-il une force plus faible au début de l’expérience ? S’il n’y a pas de sélection à priori, les différents groupes de souris (traités et non traités) devraient avoir la même force. What should we consider about all this? Maybe we should ask why scientists are searching for new drugs instead of focusing on compounds of drugs that have already shown some effects. Perhaps everyone wants to get rich, so they avoid exploring drugs that can't be patented.
What should we consider about all this? Maybe we should ask why scientists are searching for new drugs instead of focusing on compounds of drugs that have already shown some effects. Perhaps everyone wants to get rich, so they avoid exploring drugs that can't be patented. Les progrès réalisés dans la chimie médicinale des ASO ont amélioré leur profil pharmacodynamique, permettant ainsi une administration sûre et efficace au système nerveux central. Les thérapies ASO pour la SLA se sont rapidement développées au cours des deux dernières décennies, et les ASO ciblant SOD1, C9orf72 et ATXN2 sont actuellement en essais cliniques pour les formes familiales ou sporadiques de SLA.
Les progrès réalisés dans la chimie médicinale des ASO ont amélioré leur profil pharmacodynamique, permettant ainsi une administration sûre et efficace au système nerveux central. Les thérapies ASO pour la SLA se sont rapidement développées au cours des deux dernières décennies, et les ASO ciblant SOD1, C9orf72 et ATXN2 sont actuellement en essais cliniques pour les formes familiales ou sporadiques de SLA. Identifié par criblage in vitro, l'ASO ION363 développé par la société IONIS qui a aussi développé Tofersen, cible le 6e intron de FUS (SLA avec une mutation P525L).
ION363 réduit les taux de protéines de liaison à l'ARN insolubles et insolubles associées aux agrégats, telles que hnRNPA1 et ralentit la neurodégénérescence des motoneurones lombaires et la perte d'innervation de la jonction neuromusculaire.
Identifié par criblage in vitro, l'ASO ION363 développé par la société IONIS qui a aussi développé Tofersen, cible le 6e intron de FUS (SLA avec une mutation P525L).
ION363 réduit les taux de protéines de liaison à l'ARN insolubles et insolubles associées aux agrégats, telles que hnRNPA1 et ralentit la neurodégénérescence des motoneurones lombaires et la perte d'innervation de la jonction neuromusculaire. The researchers specifically focused on molecules that could reduce or reverse stress granule formation, particularly those that act directly on stress granule proteins and may be useful as therapeutic agents. From the initial screening, lipoamide emerged as a novel, potent modulator of stress granules.
Once lipoamide was identified as a hit, the researchers sought to determine its effects in cells regarding specificity, potency, intracellular localization, and its effects on other cellular condensates.
The researchers specifically focused on molecules that could reduce or reverse stress granule formation, particularly those that act directly on stress granule proteins and may be useful as therapeutic agents. From the initial screening, lipoamide emerged as a novel, potent modulator of stress granules.
Once lipoamide was identified as a hit, the researchers sought to determine its effects in cells regarding specificity, potency, intracellular localization, and its effects on other cellular condensates.