Every day brings its share of scientific articles announcing the imminent arrival of drugs for neurodegenerative diseases.
However, we are unable even to diagnose these diseases with certainty. The diagnosis is made by exclusion and sometimes gives rise to several different diagnoses. We would be better off talking about the spectrum of neurogenerative diseases. The only thing we know for certain is that these diseases are characterized by malformed protein aggregates in inappropriate places in cells.
These diseases are currently differentiated by scientists by the type of protein involved, but in fact, all of these malformed proteins are present to varying degrees in all of these diseases. The recent trend to generalize diagnosis based on molecular markers only recognizes our incompetence and only serves the pharmaceutical industry.
Amyotrophic lateral sclerosis (ALS) and/or frontotemporal dementia (FTD) are most often characterized by the cytoplasmic deposition of nuclear TAR-binding protein 43 (TDP-43). But this is rarely of a rare and deleterious protein form. Although the cytoplasmic localization of TDP-43 aggregates is commonly associated with ALS/FTD, it is unknown what causes the dysfunction, although different hypotheses have been posed such as cellular stress, for example (but not only) due to to a significant change in metabolism.
In a recent article, scientists from Macquarie University in Australia reported their work concerning the interaction between the proteins 14-3-3θ and TDP-43, which regulates the nuclear-cytoplasmic shuttle.
The 14-3-3θ protein, like many other proteins, is associated with several neurodegenerative diseases.
Similar work was carried out in the past (2016) which consisted of creating a peptide by attaching the M1 section of TDP-43 to a TAT peptide which gives a peptide: YGRKKRRQRRRAQFPGACGL, which repatriates the aggregates poorly localized in the nucleus of the cells.
This work does not seem to have given rise to recent developments in the field of neurodegenerative diseases. In any case, this 2016 work did not explain why these aggregates appeared. They only provided a mechanism to get them back into the cell's nucleus. It is not clear how this would have formed them correctly since this happens in a cellular organ called "endoplasmic reticulum" which is located in the cytoplasm.
In addition, forming proteins requires energy, but we know that the cells of many patients are in a state of "hibernation" called the cellular stress response, with activity reduced to the minimum necessary to survive. Furthermore, any genetic therapy only "infects" a fraction of cells, which reduces its interest. And these genetic therapies are not without side effects.
This new article presents a slightly different mechanism but does not further answer the questions above. The 14-3-3θ protein belongs to a family of proteins called 14-3-3, known to regulate other proteins by binding to them and they play a role in various cellular processes, including signaling, survival, and cell differentiation. This family includes more than 200 members. The authors found that neuronal levels of 14-3-3θ were increased in mouse models of ALS and sporadic FTD with TDP-43 pathology. As we already know, 14-3-3θ is associated with several neurodegenerative diseases. Scientists believe that the interaction of deleterious TDP-43 alleles with the 14-3-3θ protein results in cytoplasmic accumulation, insolubility, phosphorylation, and fragmentation of TDP-43, which resembles the pathological changes caused by these diseases in humans.
 What is interesting is that 14-3-3θ seems to interact preferentially with pathogenic TDP-43 versions but not with the usual version of TDP-43. This suggests that reducing the production (or increasing the degradation) of 14-3-3θ would reduce the production of pathogenic TDP-43. Scientists have therefore sought to reduce the amount of this 14-3-3θ protein in cells through genetic therapy.
What is interesting is that 14-3-3θ seems to interact preferentially with pathogenic TDP-43 versions but not with the usual version of TDP-43. This suggests that reducing the production (or increasing the degradation) of 14-3-3θ would reduce the production of pathogenic TDP-43. Scientists have therefore sought to reduce the amount of this 14-3-3θ protein in cells through genetic therapy.
The authors designed multiple versions of a peptide they called CTx1000, each version of which is tailored to reduce one of these deleterious forms of TDP-43. This reduction is mediated by degron of pathogenic TDP-43. A degron is a part of a protein that plays an important role in regulating protein degradation rates. In mice that underwent this gene therapy, functional deficits and neurodegeneration decreased, including when they were already symptomatic at the time of treatment. This incidentally matches many studies that, contrary to consensus, show that motor neurons do not die in ALS.
The university's press kit is, as usual, dithyrambic and the authors' statements resounding: "This new research is incredibly promising in slowing the progression of MND and FTD for the vast majority of our patients. I'm extremely hopeful that it will soon be available to our patients at the Macquarie University Hospital MND Clinic." This type of press kit is not aimed at patients and their loved ones, but rather at potential investors.
In conclusion, we can think that just as the therapy proposed in 2016 did not allow the development of a drug eight years later, it will probably be the same for this one, because it does not answer basic questions: Quid patients (the majority) who do not present a mutated form of TDP-43? What causes these protein clumps? Where can cells find the energy to be permanently “reactivated” from cellular stress response so that the therapy can do its work? How can we ensure that all of the targeted cells can receive the therapy, without side effects?
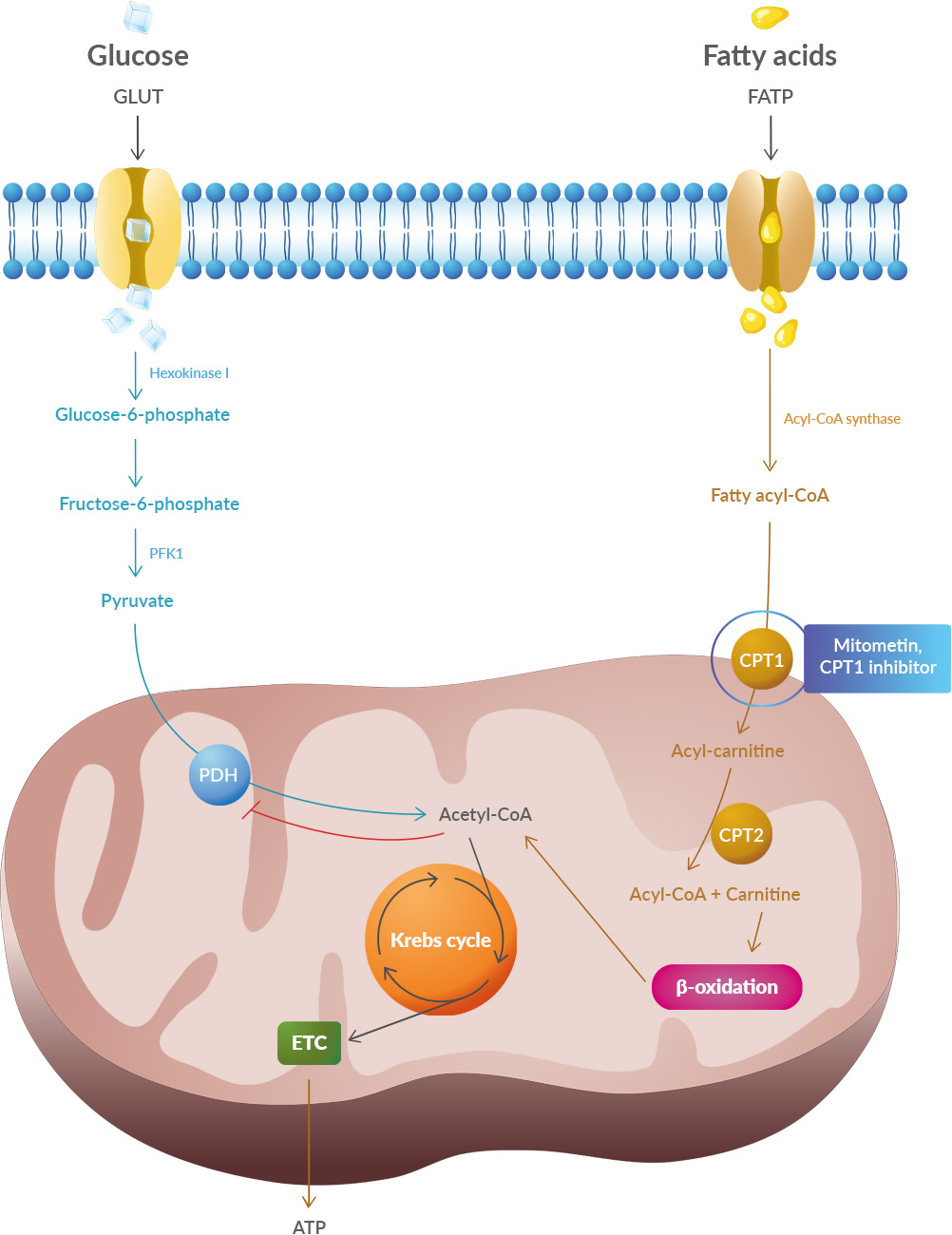 Studies have shown that oxidative stress and endoplasmic reticulum stress are correlated and can lead to protein misfolding (Abramov et al., 2020). Accumulation of misfolded proteins causes cellular damage and mitochondrial dysfunction and is associated with a range of neurodegenerative diseases, including ALS (misfolded SOD1, TDP-43, C9orf72) (McAlary et al., 2020), Parkinson's disease (misfolded α-synuclein) and Alzheimer disease (misfolded Aβ and Tau) (Abramov et al., 2020).
Studies have shown that oxidative stress and endoplasmic reticulum stress are correlated and can lead to protein misfolding (Abramov et al., 2020). Accumulation of misfolded proteins causes cellular damage and mitochondrial dysfunction and is associated with a range of neurodegenerative diseases, including ALS (misfolded SOD1, TDP-43, C9orf72) (McAlary et al., 2020), Parkinson's disease (misfolded α-synuclein) and Alzheimer disease (misfolded Aβ and Tau) (Abramov et al., 2020).
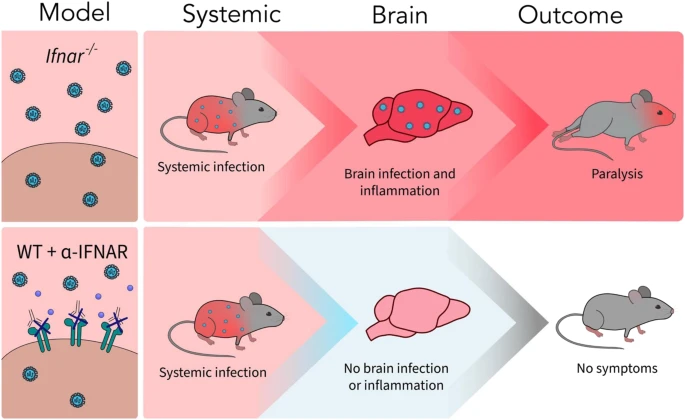 A new article aims to show that in the case of Zika viruses,
A new article aims to show that in the case of Zika viruses,  Yichang Jia, Ph.D., Co-founder of SineuGene
Yichang Jia, Ph.D., Co-founder of SineuGene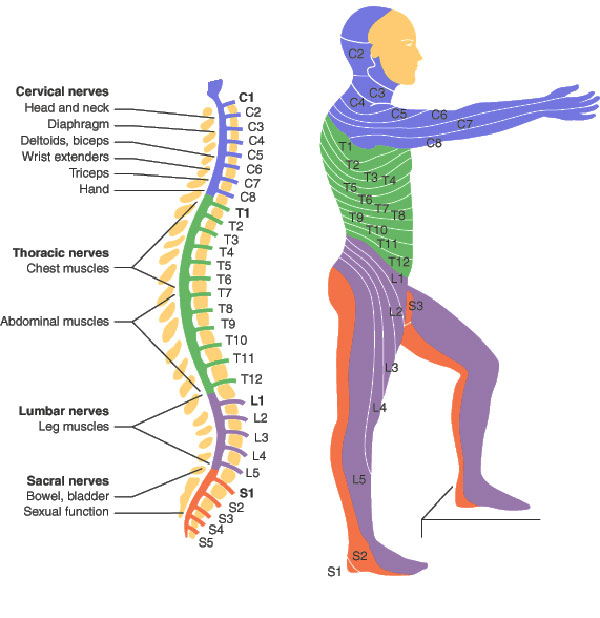 Obviously, a spinal cord injury will sever the link between the brain motor area, some upper motor neurons, and corresponding lower motor neurons and muscles. Yet it does not stop there, which is particularly interesting when we have ALS in mind. In spinal-onset ALS the disease starts in a very localized muscular for example a muscle in the thumb, and it spreads, often until respiratory muscles fail.
Obviously, a spinal cord injury will sever the link between the brain motor area, some upper motor neurons, and corresponding lower motor neurons and muscles. Yet it does not stop there, which is particularly interesting when we have ALS in mind. In spinal-onset ALS the disease starts in a very localized muscular for example a muscle in the thumb, and it spreads, often until respiratory muscles fail.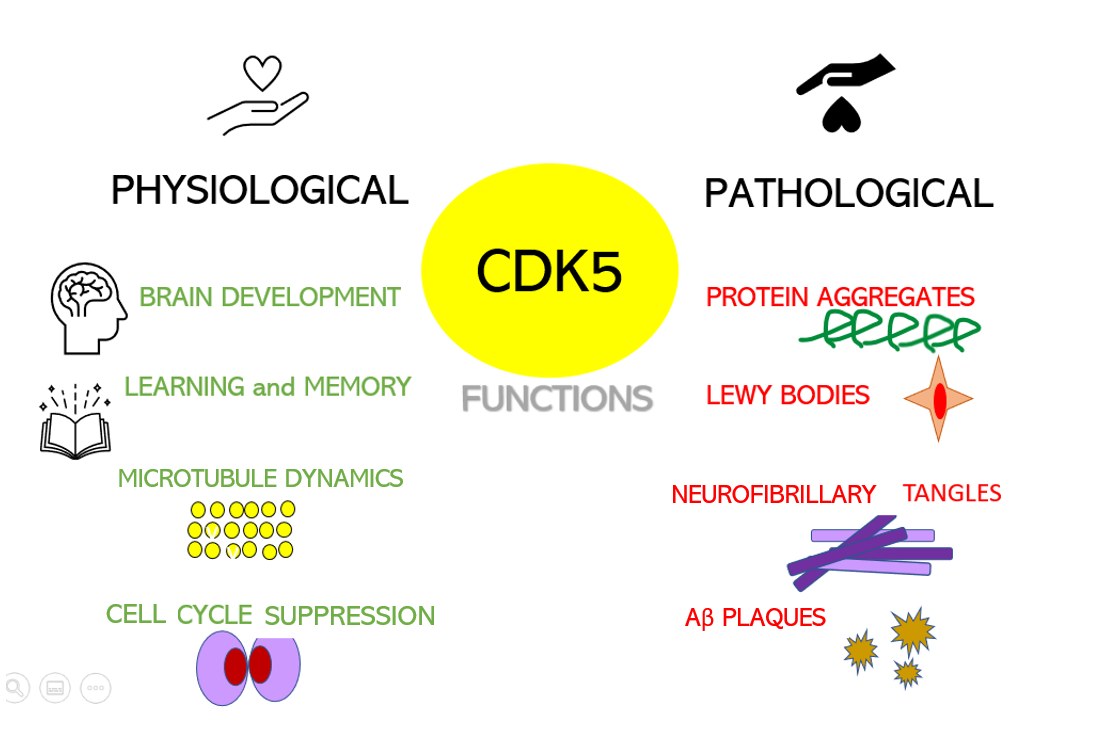 Dysregulation of CDKs, particularly cyclin-dependent kinase 5 (Cdk5), is seen in many neurological disorders, including Alzheimer's disease (AD) and Parkinson's disease (PD). Cdk5 is a unique member of the CDK family because it does not play a critical role in cell cycle progression and is not activated by a cyclin. Instead, Cdk5 is normally activated by the regulatory protein p25. Cdk5/p35/p25 activity is normally an important regulator of the proper development of the mammalian central nervous system.
Dysregulation of CDKs, particularly cyclin-dependent kinase 5 (Cdk5), is seen in many neurological disorders, including Alzheimer's disease (AD) and Parkinson's disease (PD). Cdk5 is a unique member of the CDK family because it does not play a critical role in cell cycle progression and is not activated by a cyclin. Instead, Cdk5 is normally activated by the regulatory protein p25. Cdk5/p35/p25 activity is normally an important regulator of the proper development of the mammalian central nervous system. Their results show that the model can identify, from 3 seconds of magnetoencephalographic signals, the corresponding speech segment with up to 41% accuracy, which is, however, lower than previous results. Furthermore, we know that
Their results show that the model can identify, from 3 seconds of magnetoencephalographic signals, the corresponding speech segment with up to 41% accuracy, which is, however, lower than previous results. Furthermore, we know that