Pour ce blog, je recherche plutôt des articles scientifiques récents concernant des résultats sur des humains, les essais sur des animaux modèles n'étant quasiment jamais transposables aux humains. Dans les maladies neurodégénératives il n'y a quasiment aucun essai clinique ayant montré une régression des symptômes. Actuellement il y a un fort lobbying des acteurs de l'industrie pharmaceutique et des milieux universitaires pour faire dépendre la définition des maladies de marqueurs techniques plutôt que des symptômes cliniques.
La maladie d'Alzheimer est souvent liée à l'âge, et caractérisée par une perte progressive et irréversible des neurones, à la fois des corps (soma) des neurones et des appendices comme l 'axone et les synapses. La cause de cette dégénérescence est encore inconnue, quoique l'industrie pharmaceutique et le nombre d'académiques pensent que cette destruction est due à des peptides comme les bêta-amyloïdes, ou/et la protéine Tau ou encore TDP-43, comme dans la SLA et la FTD ou encore l'alpha-synucléine comme dans la maladie de Parkinson. La majorité des études n'étudie aussi que les neurones comme si nous étions toujours en 1980. Personnellement je crois plutôt, comme l'affirme un certain nombre d'études, qu'en vieillissant nous développons un ensemble de commorbidités. Quand nous sommes arrivés à 70 ans, nous avons tous un peu de maladies d'Alzheimer et de Parkinson.
Une nouvelle étude académique semble aller dans ce sens, mais évidemment elle est faite sur des modèles de souris de la maladie et pas sur des humains.
Pour les auteurs, une certaine molécule, IDO1, provoque un cercle vicieux de déclin du soutien du métabolisme cérébral du glucose par la production de marqueurs, amyloïdes β et tau chez les sujets atteints de la maladie d'Alzheimer. Les auteurs pensent que ces marqueurs amyloïdes β et tau perturbent à leur tour le métabolisme des astrocytes et de la microglie, c'est-à-dire des cellules non neuronales qui représentent 20 % du volume total, en accroissant la production d'IDO1.
 Ces cellules, contrairement aux neurones, ressemblent plus à leurs consoeurs du reste du corps à la fois par la morphologie et la durée de vie. A l'inverse un neurone est caractérisé par un ou plusieurs appendices dendritiques et axonaux et ne se reproduisent pas. Les neurones ont besoin des astrocytes et de nombreux autres types de cellules pour survivre.
Ces cellules, contrairement aux neurones, ressemblent plus à leurs consoeurs du reste du corps à la fois par la morphologie et la durée de vie. A l'inverse un neurone est caractérisé par un ou plusieurs appendices dendritiques et axonaux et ne se reproduisent pas. Les neurones ont besoin des astrocytes et de nombreux autres types de cellules pour survivre.
Les astrocytes produisent notamment du lactate qui est exporté vers les neurones pour alimenter la respiration mitochondriale et soutenir l’activité synaptique. Ce processus est connu sous le nom de navette lactate astrocytes-neurones (ANLS). Il s'agit d'une voie métabolique par laquelle les astrocytes absorbent le glucose de la circulation sanguine, le métabolisent en lactate, puis le libèrent dans l'espace extracellulaire. Les neurones peuvent alors absorber ce lactate et l'utiliser comme source de carburant pour leurs besoins énergétiques.
Ce processus est particulièrement important pendant les périodes de forte activité neuronale, lorsque les neurones nécessitent un apport d'énergie rapide et soutenu. La monnaie énergétique des cellules est l'ATP. L'ATP est produit par les mitochondries des cellules à partir du glucose. Le glucose pénètre dans la membrane cellulaire lorsque les récepteurs cellulaires détectent l'insuline. Les neurones absorbent le glucose du sang, mais ils le font moins efficacement que les astrocytes. La navette lactate astrocyte-neurone contribue à garantir que les neurones disposent du carburant nécessaire pour maintenir leur fonction.
Les scientifiques suggèrent que l'indoleamine-2, 3-dioxygénase 1 (IDO1), une enzyme exprimée dans les astrocytes et dans de nombreux troubles neurodégénératifs, dont la maladie d'Alzheimer, est une molécule clé dans ce processus.
- L'IDO1 peut favoriser la neuroinflammation qui augmente la production de bêta-amyloïde
- L'IDO1 est une enzyme qui catalyse la dégradation de l'acide aminé tryptophane. Le tryptophane produit la sérotonine qui est un neurotransmetteur. En tant que neurotransmetteur, la sérotonine peut influencer l'activité des synapses, les jonctions entre les neurones, car les neurones libèrent davantage de neurotransmetteurs lors de la transmission synaptique, ce qui peut déclencher des voies de signalisation favorisant la production de bêta-amyloïde.
- IDO1 peut contribuer à la neuroinflammation chronique. La neuroinflammation a été associée à une augmentation de la phosphorylation de la protéine tau.
Dans le cerveau, l'IDO1 est exprimé dans les astrocytes et la microglie, ces cellules en support des neurones, mais pas dans les neurones. Bien que l'IDO1 ne participe pas directement à la voie ANLS, il joue un rôle crucial dans la régulation de l'environnement métabolique global du cerveau. La dégradation du tryptophane produit également de la kynurénine. La kynurénine peut stimuler la production de facteurs neuroprotecteurs, tels que le BDNF (facteur neurotrophique dérivé du cerveau). Ces facteurs peuvent aider à protéger les neurones contre les dommages et favoriser leur survie. En régulant les niveaux de tryptophane et de ses métabolites, IDO1 peut influencer indirectement le métabolisme équilibre du cerveau, y compris la disponibilité du glucose et du lactate.
Les auteurs rapportent que l'inhibition de l'IDO1 et la production de kynurénine sauvent la plasticité synaptique dans l'hippocampe et la fonction de mémoire dans les animaux modèles de pathologie amyloïde et tau en rétablissant le soutien métabolique astrocytaire des neurones.
Au contraire l'activation de l'IDO1 dans les astrocytes par les oligomères amyloïdes β et tau, augmenterait le kynurénine et supprime la glycolyse (la transformation du glucose en ATP) d'une manière dépendante de l'AhR.
L’inhibition pharmacologique de l’IDO1 restaurerait la glycolyse astrocytaire et la production de lactate. Chez les souris APPSwe-PS1∆E9 et 5XFAD productrices d’amyloïde et chez les souris P301S productrices de tau, l’inhibition de l’IDO1 améliore le glucose de l’hippocampe et restaure la mémoire spatiale. Cette affirmation est assez surprenante : Lors de la maladie d'Alzheimer, une partie conséquente du cerveau meurt et disparaît, comment la mémoire pourrait-elle être restituée ?
Le blocage de l’IDO1 sauve également la potentialisation hippocampique à long terme d’une manière dépendante du transporteur de monocarboxylate, ce qui suggère que l’activité de l’IDO1 perturbe le soutien métabolique astrocytaire des neurones. En effet, l’IDO1 régule la production de lactate par les astrocytes qui est ensuite absorbé par les neurones humains. Dans les cocultures d'astrocytes et de neurones issus de sujets atteints de la maladie d'Alzheimer, la production déficiente de lactate des astrocytes et son transfert vers les neurones ont été corrigés par l'inhibition de l'IDO1, ce qui a permis d'améliorer le métabolisme neuronal du glucose.
Il se trouve que des inhibiteurs de l’IDO1 pénétrants dans le cerveau ont déjà été développés comme traitement d’appoint contre le cancer, ils pourraient donc être réutilisés pour traiter des maladies neurodégénératives telles que la maladie d’Alzheimer. A tout le moins cela veut dire qu'une phase II pourrait être démarrée rapidement si un financement est trouvé.
Cette étude suggère également qu'en plus de la maladie d'Alzheimer, la manipulation de l'IDO1 peut être pertinente pour la démence de la maladie de Parkinson, qui est caractérisée par une accumulation d'amyloïdes en plus de l'α-synucléine, ainsi que pour le grand spectre des tauopathies. Il est possible qu'un métabolisme astrocytaire déficient du glucose puisse également être à l'origine d'autres maladies neurodégénératives caractérisées par l'accumulation d'autres protéines mal répondues où des augmentations des métabolites de la voie de la kynurénine ont été observées.
En effet, des états dysfonctionnels d’étapes distinctes de la voie de la kynurénine ont été décrits dans de nombreux troubles neurologiques.
 Currently, the diagnosis of Alzheimer’s disease includes cognitive decline leading to dementia, associated with the observation of two major proteinopathies in the brain tissue. These two proteinopathies are plaques, formed by the aggregation of beta-amyloid proteins, and tangles, which are composed of hyperphosphorylated tau proteins.
Currently, the diagnosis of Alzheimer’s disease includes cognitive decline leading to dementia, associated with the observation of two major proteinopathies in the brain tissue. These two proteinopathies are plaques, formed by the aggregation of beta-amyloid proteins, and tangles, which are composed of hyperphosphorylated tau proteins. Studies have shown that immune cells in the brain, particularly microglia, and astrocytes, are key regulators of the inflammatory response in the central nervous system and are involved in the pathogenesis of AD. These cells have various roles, including supporting neurons.
Studies have shown that immune cells in the brain, particularly microglia, and astrocytes, are key regulators of the inflammatory response in the central nervous system and are involved in the pathogenesis of AD. These cells have various roles, including supporting neurons.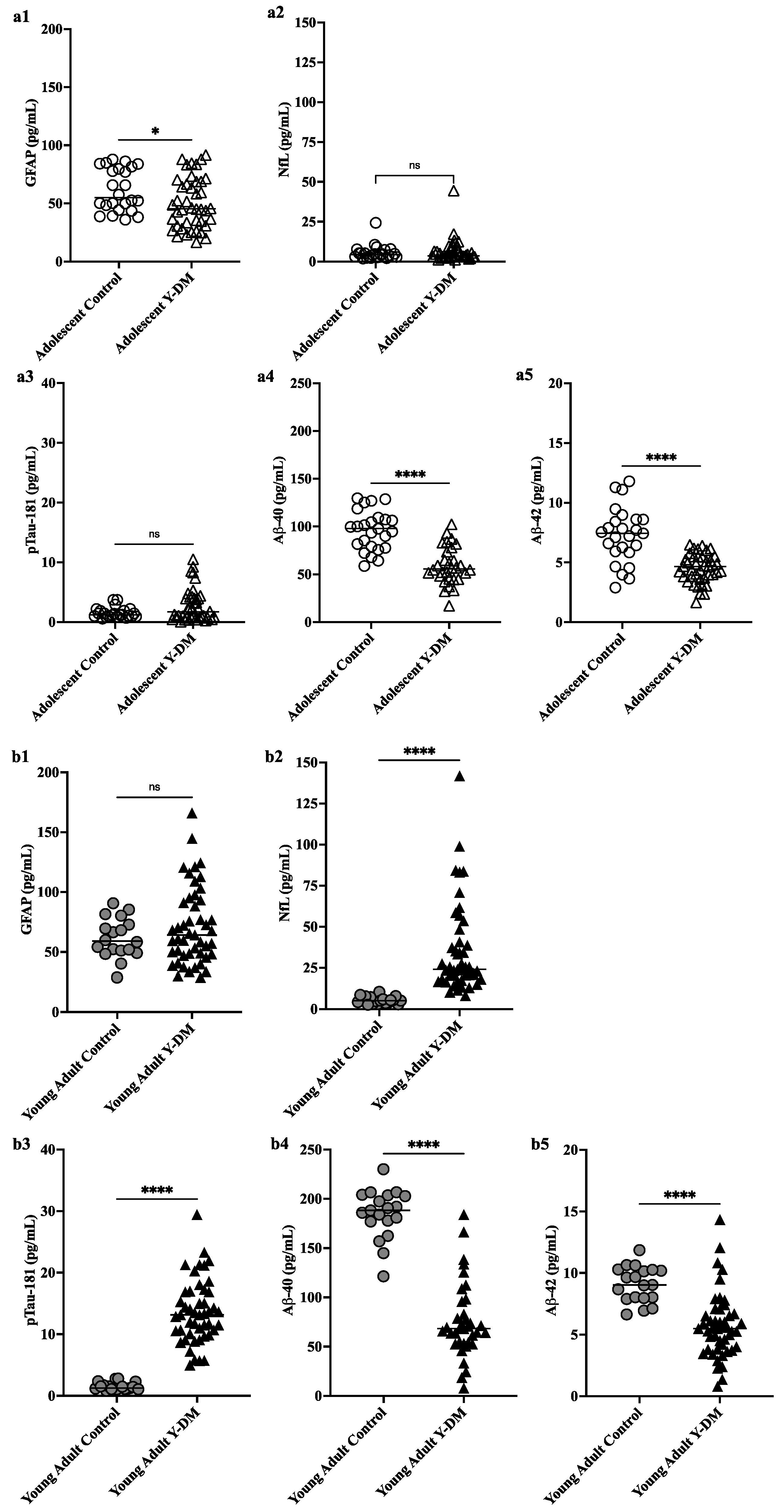 This study aimed to explore neurodegenerative disease biomarkers in cohort-derived biomarker banks as changes in key plasma biomarkers between the time of diabetes diagnosis and early adulthood have been correlated with worsening cognitive function in young adults with early-onset diabetes.
This study aimed to explore neurodegenerative disease biomarkers in cohort-derived biomarker banks as changes in key plasma biomarkers between the time of diabetes diagnosis and early adulthood have been correlated with worsening cognitive function in young adults with early-onset diabetes. Sleep efficiency in older adults. Sleep deprivation increases amyloid-β (Aβ) concentrations in the interstitial fluid of experimental animal models and in cerebrospinal fluid in humans, while increased sleep decreases Aβ. Sleep abnormalities may therefore represent a risk factor for neurodegeneration.
Sleep efficiency in older adults. Sleep deprivation increases amyloid-β (Aβ) concentrations in the interstitial fluid of experimental animal models and in cerebrospinal fluid in humans, while increased sleep decreases Aβ. Sleep abnormalities may therefore represent a risk factor for neurodegeneration. These articles provide the reader with several points for further reflection.
- One of them is that the clinical trial cannot be the only method of validating the marketing of a drug. Indeed, for diseases that develop over decades, often asymptomatically, how can the effectiveness of a drug be tested? We can always dream of a pill that will cure in a few months a patient, as this is true for diseases where there is an identified pathogen, but it is very unlikely with chronic diseases. Even for communicable diseases, this type of medication does not always exist; a patient can be considered permanently cured, even though they suffer very serious after-effects.
These articles provide the reader with several points for further reflection.
- One of them is that the clinical trial cannot be the only method of validating the marketing of a drug. Indeed, for diseases that develop over decades, often asymptomatically, how can the effectiveness of a drug be tested? We can always dream of a pill that will cure in a few months a patient, as this is true for diseases where there is an identified pathogen, but it is very unlikely with chronic diseases. Even for communicable diseases, this type of medication does not always exist; a patient can be considered permanently cured, even though they suffer very serious after-effects. The exact reasons why atrial fibrillation (a common heart disease) increases dementia risk aren't fully understood, but there are likely several factors involved. Some of these factors directly cause damage, while others increase the chances of getting dementia over time. These factors include tiny blood blockages and bleeding in the brain's small blood vessels. Small strokes you might not even notice, reduce blood flow to the brain, increase long-term inflammation, and lead to shrinkage of brain tissue.
The exact reasons why atrial fibrillation (a common heart disease) increases dementia risk aren't fully understood, but there are likely several factors involved. Some of these factors directly cause damage, while others increase the chances of getting dementia over time. These factors include tiny blood blockages and bleeding in the brain's small blood vessels. Small strokes you might not even notice, reduce blood flow to the brain, increase long-term inflammation, and lead to shrinkage of brain tissue.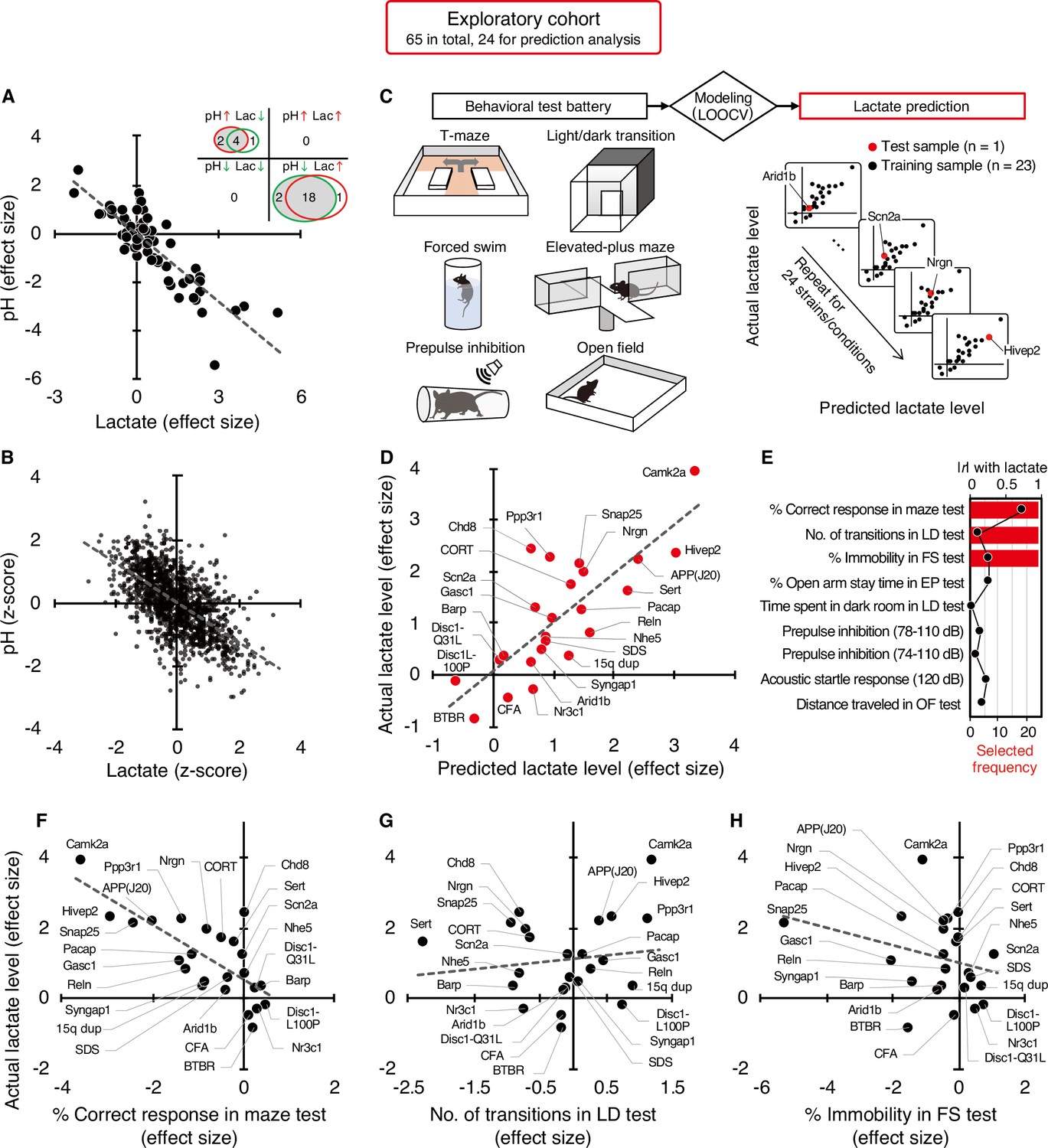 What could be the mechanism between increased lactate and decreased pH?
Lactate is a byproduct of metabolism and it is an acid, hence the acidification of pH. Neuronal activity relies heavily on glucose for energy. Under normal conditions, oxygen is readily available, and glucose is broken down through aerobic respiration, producing ATP (energy) with minimal lactate as a byproduct. However, during periods of high neuronal activity or limited oxygen supply, cells shift to anaerobic metabolism, relying more on glycolysis. This process produces more lactate as a byproduct.
What could be the mechanism between increased lactate and decreased pH?
Lactate is a byproduct of metabolism and it is an acid, hence the acidification of pH. Neuronal activity relies heavily on glucose for energy. Under normal conditions, oxygen is readily available, and glucose is broken down through aerobic respiration, producing ATP (energy) with minimal lactate as a byproduct. However, during periods of high neuronal activity or limited oxygen supply, cells shift to anaerobic metabolism, relying more on glycolysis. This process produces more lactate as a byproduct. Improving cognition using a simple probiotic would be an extraordinary result due to its simplicity of implementation.
Improving cognition using a simple probiotic would be an extraordinary result due to its simplicity of implementation.