This article is a bit unusual as it claims a relationship between several genes in ALS (Lou Gerigh disease), including APP which is associated with Alzheimer's disease. On the overall it says that C9 ALS is a problem of ribosome quality control (RQC) which leads to cellular stress response. It suggests that C9, FUS, TDP-43 mutations influence mTORC2 protein which in turn alters the translation mechanism. When the translation mechanism is altered, cellular stress response is activated and protein production nearly stops, rendering the cell non-functional. It's the start of the disease.
ALS is a muscle wasting disease characterized by degeneration of lower motor neurons and axons and loss of upper motor neurons and their corticospinal tracts. FTD is a progressive neuronal atrophy with neuronal loss in the frontal and temporal cortices and associated behavioral and personality changes and impairment of language skills. Advances in human genetics have identified multiple genetic mutations commonly associated with ALS and FTD, revealing that these two diseases are related and may represent a continuum of a broad neurodegenerative disorder. C9orf72 mutation is present in approximately 40% of familial ALS and 8-10 % of sporadic ALS. It is currently the most common demonstrated mutation related to ALS - far more common than SOD1.
The research on association of C9ORF72 with ALS or FTD is relatively recent and its mechanisms are not clear. This obviously impairs the proposal of new drugs. Gene expression or translation is the process in which ribosomes in the cytoplasm or endoplasmic reticulum synthesize proteins after the process of transcription of DNA to RNA in the cell's nucleus. Yet during translation elongation, ribosomes may slowdown or even stall for various reasons. The polypeptide later folds into an active protein and is sent to its final location to perform its functions in the cell.
RAN translation, is an irregular mode of mRNA translation that can occur in eukaryotic cells. RAN translation produces a variety of dipeptide repeat proteins (DPR) by translation of expanded hexanucleotide repeats present in an intron of the C9orf72 gene. The expansion of the hexanucleotide repeats and thus accumulation of dipeptide repeat proteins are thought to cause cellular toxicity that leads to neurodegeneration in ALS disease.
Previous studies of protein quality control have focused on how proteins were handled after translation. However, rproblems with proteostasis are prevalent even with translating nascent peptide chains still associated with ribosomes, necessitating ribosome-associated quality control (RQC) mechanisms.
Mutations in other genes that are commonly linked to ALS/FTD have also shed lights on disease pathogenesis such as TDP-43 and FUS. Other genes linked to ALS/FTD include VCP, a member of the AAA ATPase family with established function in the recycling and degradation of ubiquitinated proteins, and genes with functions in protein clearance or maintenance of protein homeostasis. In addition, upregulation of APP, a protein whose aberrant processing or metabolism having been implicated in Alzheimer’s disease (AD), was observed at early stages of ALS and FTD, presumably as a compensatory response to neuronal damage or impairment of axonal transport. However, the relationships among the various ALS/FTD genes remain underexplored.
It is therefore important to understand cellular mechanisms underlying the quality control of poly(GR). Previous studies of protein quality control have focused on how proteins were handled after translation, e.g., by chaperone-mediated refolding, or proteasome- and lysosome-mediated degradation. However, recent studies reveal that problems with proteostasis are prevalent even with translating nascent peptide chains still associated with ribosomes, necessitating ribosome-associated quality control (RQC) mechanisms.
In the case of poly(GR), it was shown that its translation was frequently stalled, presumably due to positively charged arginine residues interacting with negatively-charged residues lining the exit tunnel of 60S ribosome. Stalled poly(GR) translation activates the RQC process, the inadequacy of which can lead to the accumulation of aberrant, C-terminally modified (CAT-tailed) poly(GR) species that can perturb proteostasis and contribute to poly(GR) accumulation and neuromuscular degeneration.
In this study, the scientists from USA and China set out to test whether the other ALS/FTD associated genes may participate in the quality control of poly(GR). Strikingly, they discovered that overexpression of APP, FUS, and TDP-43 restrains poly(GR) protein expression. Mechanistically, APP, FUS and TDP-43 act through the mTORC2/AKT/VCP axis to regulate the RQC of poly(GR) translation. Inhibition of the mTORC2/AKT/VCP axis could restore poly(GR) protein expression attenuated by APP, FUS, or TDP-43. Their data strongly implicate the mTORC2/AKT/VCP axis as a major regulator of protein quality control in ALS/FTD.
Their data support the working model that mutated APP, FUS, and TDP-43 are upstream regulators of the mTORC2/AKT/VCP axis, which regulates the RQC of poly(GR) during its translation stalling. Moreover, they suggest that APP, FUS, and TDP-43 can also induce repression of global translation when ribosome stalling is persistent.
APP acts through the mTORC2/AKT signaling axis to regulate the RQC of C9-ALS/FTD-associated poly(GR) translation. The involvement of APP in ALS has previously been studied in the context of ALS, and APP or its metabolite was found to exacerbate ALS-related phenotypes in the SOD1-G93A mouse model. This new results suggest that APP can activate mTORC2/AKT signaling to alleviate stalled translation of poly(GR) and restrain the expression of aberrant poly(GR) translation products, at least at the initial stage. It is possible that in ALS/FTD setting, APP is upregulated as a protective response in response to neuronal damage at an early stage of disease as previously suggested
It is difficult to not think about the controverse about the role of amyloid plaques found in the brains of Alzheimer's disease patients. Amyloid beta is a fragment from the larger amyloid-beta precursor protein (APP) a transmembrane protein that penetrates the neuron's membrane. APP is critical to neuron growth, survival, and post-injury repair.
While the authors write only about ALS, chronic upregulation of APP may contribute not only to ALS, but also also to Alzheimer's disease due to the accumulation of APP metabolites, the stalled translation of APP itself, or the prolonged activation of stress response pathways by APP may lead to the depression of global translation.
Indeed the authors found that integrated stress response as indicated by eIF2α phosphorylation was heightened in transgenic flies expressing poly(GR). This is presumably caused by the ribosome stalling occurring during poly(GR) translation.
A similar situation may occur with TDP-43 and FUS. In fact, both mutations in TDP-43 and FUS genes have been shown to associate with stalled ribosomes, and in the case of TDP-43, its association with stalled ribosomes provides neuroprotection function in the face of sublethal stress.
Intriguingly, the authors showed that the a portion of APP (APP-C99) is sufficient to activate the mTORC2/AKT axis and regulate GR80 translation, whereas the Aβ-42 portion of APP was without effect. This finding resonates with recent revelation of aberrant APP-C99 as the etiological driver of Alzheimer’s disease.
Remarkably, the translation of this portion of APP is also frequently stalled, the inadequate RQC of which can generate aberrant translation products that precipitate hallmarks of Alzheimer’s disease. It is therefore fascinating that overexpression one stalled translation product (APP-C99) would abrogate the stalled translation of another portion (GR-80).
Future studies will investigate at the biochemical level how APP/APP-C99, FUS, and TDP-43 signal to the mTORC2/AKT/VCP axis to regulate the RQC of stalled poly(GR) translation, whether endogenous stalled peptides that serve as RQC substrate(s) may also targeted by this pathway, and how this signaling process may be targeted for therapeutic purposes.

 NLRP3 is expressed predominantly in macrophages and as a component of the inflammasome, it detects products of damaged cells such as extracellular ATP and crystalline uric acid. Activated NLRP3 in turn triggers an immune response. Mutations in the NLRP3 gene are associated with a number of organ specific autoimmune diseases.
NLRP3 is expressed predominantly in macrophages and as a component of the inflammasome, it detects products of damaged cells such as extracellular ATP and crystalline uric acid. Activated NLRP3 in turn triggers an immune response. Mutations in the NLRP3 gene are associated with a number of organ specific autoimmune diseases. Curiously Mice exposed to high-dose radiation (2 Gy)did not show these effects and associations. Intriguily, there are also reports indicating stimulatory or beneficial effects after exposure to cell phone radiofrequency radiation.
Curiously Mice exposed to high-dose radiation (2 Gy)did not show these effects and associations. Intriguily, there are also reports indicating stimulatory or beneficial effects after exposure to cell phone radiofrequency radiation. For sedentary adults, basal metabolic rate (the metabolic rate at rest) accounts for about 50% to 70% of total energy output, dietary thermogenesis for 10% to 15%, and physical activity for the remaining 20% to 30%.
For sedentary adults, basal metabolic rate (the metabolic rate at rest) accounts for about 50% to 70% of total energy output, dietary thermogenesis for 10% to 15%, and physical activity for the remaining 20% to 30%.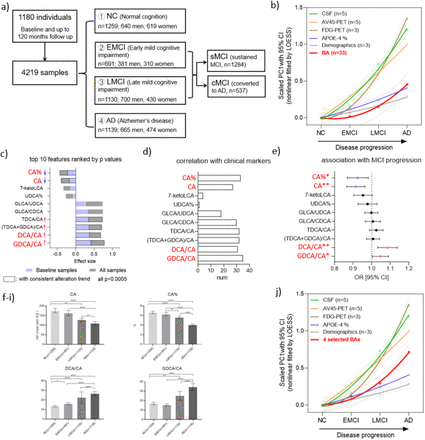
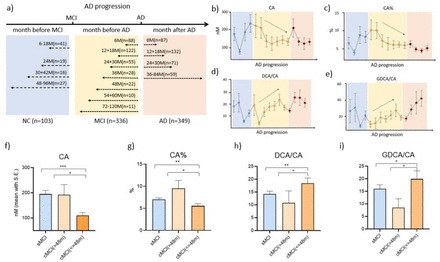
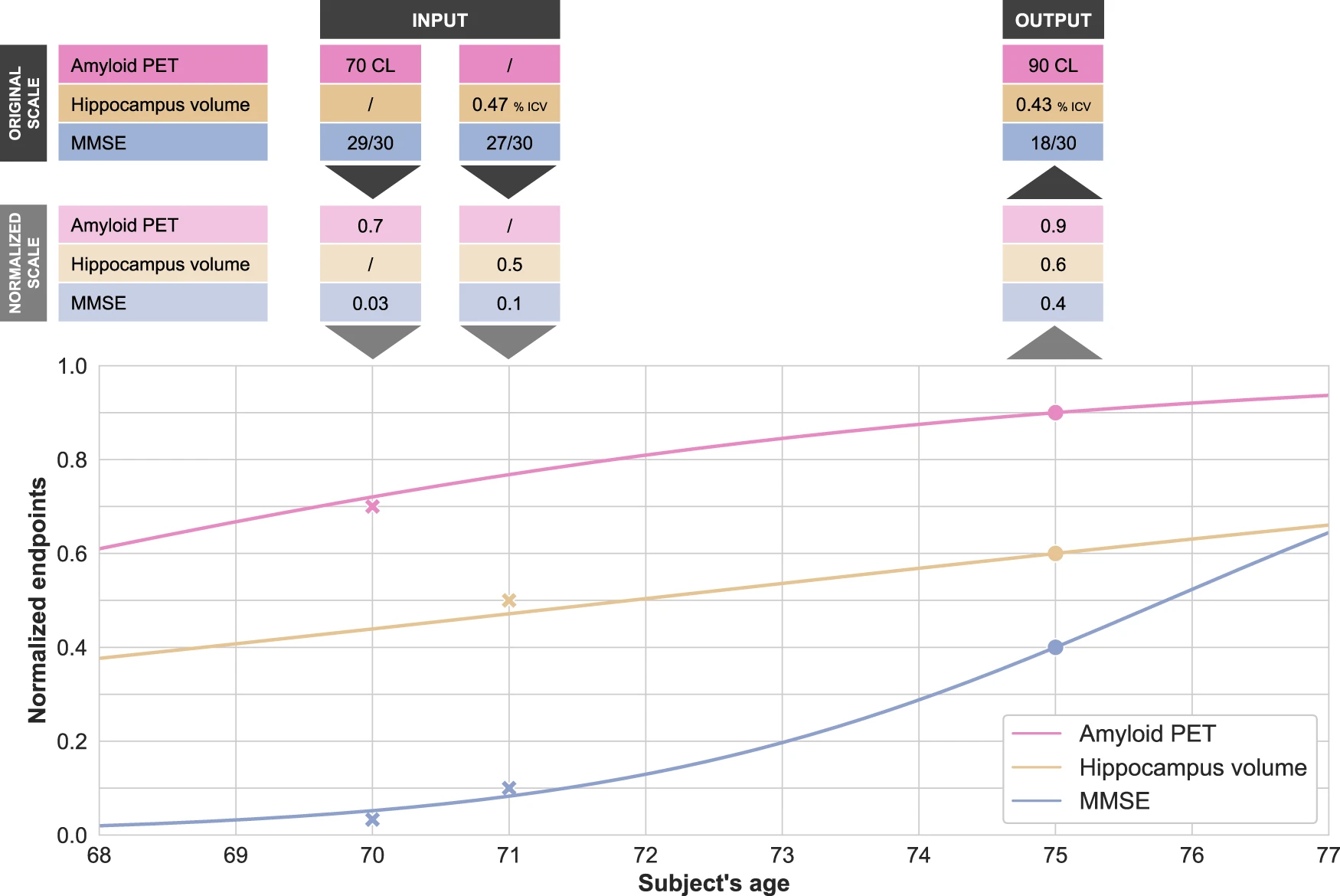 Disease modeling uses computational and statistical methods to address this question. These models learn the variability of disease progression from observational longitudinal cohort data and can then predict the progression of patients from their historical data. They require various clinical or biomarker assessments at one or several time points as input.
Disease modeling uses computational and statistical methods to address this question. These models learn the variability of disease progression from observational longitudinal cohort data and can then predict the progression of patients from their historical data. They require various clinical or biomarker assessments at one or several time points as input. The essential step for tau protein aggregation is tau phosphorylation which may also play a role in initiating β-amyloid toxicity. One of the kinases that phosphorylate tau protein is glycogen synthase kinase-3 beta (GSK3β); insulin signaling inhibits the activity of this kinase. Therefore, a hypothesis suggests that GSK3β deregulation in neurons may be a key point in developing Alzheimer’s disease.
The essential step for tau protein aggregation is tau phosphorylation which may also play a role in initiating β-amyloid toxicity. One of the kinases that phosphorylate tau protein is glycogen synthase kinase-3 beta (GSK3β); insulin signaling inhibits the activity of this kinase. Therefore, a hypothesis suggests that GSK3β deregulation in neurons may be a key point in developing Alzheimer’s disease.