Le sommeil est important pour maintenir la santé du cerveau.
Pendant le sommeil, le cerveau élimine les déchets métaboliques, les protéines et les débris cellulaires qui s'accumulent pendant l'activité cérébrale diurne.
Ceet élimination se fait via grâce à des systèmes interconnectés d'activité neuronale et de flux de liquide interstitiel et céphalo-rachidien.
 Des études précliniques ont montré que pendant le sommeil, l'infiltration du liquide interstitiel et céphalo-rachidien le long des espaces périvasculaires augmente, augmentant ainsi la clairance des solutés interstitiels. Lorsqu'ils sont agrandis, les espaces périvasculaires sont visibles par imagerie par résonance magnétique cérébrale (IRM).
Le volume et le nombre de espaces périvasculaires augmentent avec l'âge.
Ce phénomène est associés à des facteurs de risque vasculaire tels que l'hypertension, des marqueurs de microangiopathie telle que la leucoaraiose et entraine des effets néfastes sur la santé. Une question importante est de savoir si le manque de sommeil est la cause de ces risques vasculaires, ou simplement leur conséquence.
Des études précliniques ont montré que pendant le sommeil, l'infiltration du liquide interstitiel et céphalo-rachidien le long des espaces périvasculaires augmente, augmentant ainsi la clairance des solutés interstitiels. Lorsqu'ils sont agrandis, les espaces périvasculaires sont visibles par imagerie par résonance magnétique cérébrale (IRM).
Le volume et le nombre de espaces périvasculaires augmentent avec l'âge.
Ce phénomène est associés à des facteurs de risque vasculaire tels que l'hypertension, des marqueurs de microangiopathie telle que la leucoaraiose et entraine des effets néfastes sur la santé. Une question importante est de savoir si le manque de sommeil est la cause de ces risques vasculaires, ou simplement leur conséquence.
La microangiopathie est une angiopathie qui atteint des vaisseaux sanguins de petits calibres. Elle est souvent une complication du diabète et de l'hypertension artérielle. La présence de leucoaraiose sur l'IRM a également été associée à un déclin fonctionnel, à des troubles de la marche et à la dépression, peut-être parce qu'ils perturbent les réseaux neuronaux.

Souvent, les leucoaraiose sont dus à un processus appelé infarctus incomplet, c'est à dire une réduction chronique du flux sanguin dans les zones profondes du cerveau causée par l'artériolosclérose, la lipohyalinose ou la nécrose fibrinoïde des petites artères et artérioles cérébrales. Un tel flux sanguin réduit conduit à l'hypoxie, altère les mécanismes d'autorégulation cérébrale et favorise la transcription des gènes inflammatoires, la rupture de la barrière hémato-encéphalique et l'entrée de protéines pro-inflammatoires dans les parois des vaisseaux et le parenchyme cérébral. Même en l'absence d'infarctus franc, ces processus entraînent une démyélinisation, une perte axonale, une réduction de la densité gliale, une vacuolisation et une atrophie du cortex sus-jacent.
En outre, d'autres processus pouvant être impliqués dans la genèse des leucoaraiose comprennent le dysfonctionnement des cellules précurseurs des oligodendrocytes, la défaillance du système glymphatique, la collagénose veineuse. Les études IRM qui utilisent des techniques telles que l'imagerie du tenseur de diffusion et le transfert d'aimantation pour examiner la diffusivité de l'eau et l'intégrité de la substance blanche montrent que certains des changements physiopathologiques précoces se produisent également dans les zones de la substance blanche qui semblent normales sur l'IRM conventionnelle. Ces études IRM des études suggèrent que les leucoaraiose visibles ne sont «que la pointe de l'iceberg» et que la physiopathologie sous-jacente est un processus diffus affectant les petits vaisseaux sanguins dans une grande partie de la substance blanche et d'autres parties du cerveau.
Les marqueurs de la qualité du sommeil ont été associés à plusieurs indicateurs structurels de la détérioration de la santé cérébrale, à savoir les hyperintensités de la substance blanche (Leucoaraiose) d'origine vasculaire présumée, les microhémorragies et la perte de tissu cérébral d'apparence normale, tous évalués à l'aide IRM.
Cependant, les études démontrant des associations causales définitives entre le sommeil et la santé des tissus cérébraux au fil du temps chez les personnes âgées vivant dans la communauté font défaut.
Des scientifiques Ecossais essayent de répondre à cette question dans une nouvelle publication, pour cela ils utilisent les données longitudinales des personnes âgées vivant dans la communauté de la Lothian Birth Cohort 1936. Les participants à l'étude sont donc des personnes âgées toutes nées en 1936 à Édimbourg et dans les environs (Lothian). Le Lothian est une région d'Écosse où la langue des Lothiens et de l'ancien royaume de Northumbria est encore pratiquée.
La Lothian Birth Cohort 1936 est une vaste étude basée sur la population du vieillissement cognitif, cérébral et général. Les auteurs se sont intéressés à toute association entre la durée, la qualité et l'efficacité du sommeil autodéclarées, la charge espaces périvasculaires au début de la 8e décennie de la vie et la détérioration des volumes cérébraux normaux et anormaux au cours de la décennie, à l'aide de l'IRM structurelle.
Là où des associations existent, les scientifiques ont cherché à savoir si la charge espaces périvasculaires influence ou non l'association entre le sommeil et les changements structurels du cerveau sur une période de temps.
Ils ont testé trois hypothèses:
(1) les mesures du sommeil serait associées indépendamment à une détérioration ultérieure du tissu cérébral,
(2) la charge espaces périvasculaires serait associée à une détérioration du tissu cérébral,
(3) l'association entre le sommeil et les changements structurels du cerveau serait en partie médiée par le espaces périvasculaires fardeau.
Dans ce grand échantillon d'individus, le fardeau des espaces périvasculaires élargies visibles dans l'examen IRM du cerveau à l'âge d'environ 73 ans était associé à une aggravation ultérieure de la santé de la substance blanche. Celle-ci était reflétée par une diminution plus rapide du volume de substance blanche d'apparence normale, une plus grande augmentation de leucoaraiose et de la «mesure des dommages à la substance blanche» des années 73 à 79.
Les personnes qui ont signalé une moins bonne efficacité du sommeil et plus de sommeil diurne étaient celles qui ont également connu une baisse significativement plus importante de la santé de leur substance blanche entre 73 et 79 ans, et leurs résultats montrent que cela pourrait être en partie médié par la charge espaces périvasculaires.
Ni la charge espaces périvasculaires, ni la qualité du sommeil ou les indicateurs d'efficacité n'ont été associés à une détérioration du volume de tissue cérébral ou des changements de matière grise au cours de la même période.
Une analyse longitudinale récente à 5 points dans le temps sur 28 ans dans la même cohorte avait auparavant identifié quatre groupes de trajectoires différents avec des durées moyennes de sommeil différentes allant d'environ 5 à 8 h, mais n'avait trouvé aucune différence significative dans la microstructure de la matière blanche ou les volumes de matière grise entre les groupes.
Une autre étude à la fin de l'âge adulte [60–82 ans] avait déjà révélé que le nombre de fois où les participants ont signalé une mauvaise qualité de sommeil sur cinq points temporels couvrant une période de 16 ans avant l'analyse, n'était pas associé à ces mesures de la substance blanche, suggérant ainsi que seule la qualité actuelle du sommeil affectait la substance blanche.
Avec cette étude, les résultats peuvent indiquer soit un effet à court terme du manque d'efficacité du sommeil sur la santé de la substance blanche à la fin de l'âge adulte, soit que l'altération des habitudes de sommeil chez les personnes âgées peut être liée à des changements cérébraux sous-jacents mais ne provoque pas nécessairement les changements cérébraux.
Bien que l'association entre les espaces périvasculaires élargi et le leucoaraiose ait été rapportée, l'étude actuelle suggère qu'en outre, une augmentation des espaces périvasculaires visible (en volume, en nombre ou en scores visuels) prédit également le taux d'atrophie de la substance blanche et la structure de la substance blanche.
dommages à la fin de l'âge adulte, mais pas de changements de volume total du cerveau et de la matière grise.
Les troubles du sommeil sont associés à une détérioration plus rapide de la santé de la substance blanche chez les septuagénaires.
Une charge plus élevée des espaces périvasculaires est associée à une détérioration plus rapide de la santé de la substance blanche chez les septuagénaires.
Les espaces périvasculaires interviennent en partie dans l'effet du sommeil sur la santé du cerveau.
Le principal apport de cette étude semble donc être que c'est la détérioration de la santé vasculaire, plutôt que le manque de sommeil, qui amène la détérioration de la matière blanche.
Cependant, il faut faire preuve de prudence en ce qui concerne le manque apparent d'association avec la matière grise et les taux d'atrophie totale du tissu cérébral.
Le logiciel utilisé pour générer des volumes de matière grise, FSL-FAST, bien que considéré comme un étalon-or pour la segmentation des tissus d'apparence normale, ne discrimine pas la matière grise saine de la matière grise malsaine.
L'absence d'association entre les paramètres du sommeil et l'atrophie de la matière grise est moins surprenante.
Une étude précédante examinant la qualité et la quantité du sommeil en relation avec les changements corticaux avait indiqué que la qualité du sommeil et les troubles du sommeil autodéclarés étaient liés à l'amincissement du cortex temporal latéral droit.
Aucune information sur l'apnée du sommeil n'a été recueillie, mais dans les dossiers médicaux, elle n'a été signalée que chez un seul participant.
 Telomerase reverse transcriptase (abbreviated to TERT, or hTERT in humans) is a subunit of the enzyme telomerase.
TERT inhibition has been linked directly or indirectly to all hallmarks of aging, but TERT expression is linked to the development of many cancers.
Unfortunately, TERT gene is epigenetically repressed with the onset of aging markers in all tissues.
Telomerase reverse transcriptase (abbreviated to TERT, or hTERT in humans) is a subunit of the enzyme telomerase.
TERT inhibition has been linked directly or indirectly to all hallmarks of aging, but TERT expression is linked to the development of many cancers.
Unfortunately, TERT gene is epigenetically repressed with the onset of aging markers in all tissues. Ribosomopathies encompass a wide range of syndromes. Common symptoms include a reduced number of blood cells, predisposition to cancer, skeletal abnormalities, and failure to thrive. Ribosomopathy has been associated with skeletal muscle atrophy, which brings us closer to ALS.
Ribosomopathies encompass a wide range of syndromes. Common symptoms include a reduced number of blood cells, predisposition to cancer, skeletal abnormalities, and failure to thrive. Ribosomopathy has been associated with skeletal muscle atrophy, which brings us closer to ALS. There is nothing new in the list of drugs the authors list, and they lump in the same basket many unrelated drugs.
There is nothing new in the list of drugs the authors list, and they lump in the same basket many unrelated drugs. Curtiss Neveu via Wikipedia
Curtiss Neveu via Wikipedia Les lymphocytes T matures sont considérés comme immunologiquement naïfs jusqu'à ce qu'ils rencontrent le peptide spécifique dans le contexte d'une molécule d'antigène leucocytaire humain (HLA) que leur récepteur reconnaît. Une fois la reconnaissance de l’antigène effectuée, les cellules reçoivent un signal prolifératif qui conduit à une expansion marquée des lymphocytes T spécifiques de l’antigène et à une réponse inflammatoire. Bien que beaucoup de ces cellules subissent l’apoptose après la réponse initiale, d’autres sont sauvées de la rétraction immunitaire et persistent sous forme de cellules T mémoire. Les lymphocytes T mémoire peuvent répondre rapidement à une nouvelle provocation spécifique d’un antigène et persister dans la circulation à long terme
Les lymphocytes T matures sont considérés comme immunologiquement naïfs jusqu'à ce qu'ils rencontrent le peptide spécifique dans le contexte d'une molécule d'antigène leucocytaire humain (HLA) que leur récepteur reconnaît. Une fois la reconnaissance de l’antigène effectuée, les cellules reçoivent un signal prolifératif qui conduit à une expansion marquée des lymphocytes T spécifiques de l’antigène et à une réponse inflammatoire. Bien que beaucoup de ces cellules subissent l’apoptose après la réponse initiale, d’autres sont sauvées de la rétraction immunitaire et persistent sous forme de cellules T mémoire. Les lymphocytes T mémoire peuvent répondre rapidement à une nouvelle provocation spécifique d’un antigène et persister dans la circulation à long terme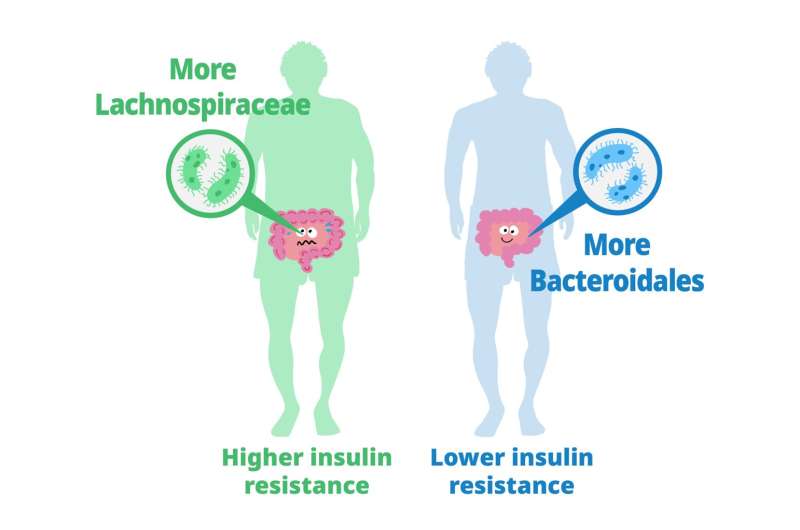 Previous studies have explored the role of gut microbiota in metabolizing nutrients in insulin resistance. This research aims to uncover the mechanisms underlying this relationship using a multi-omics approach. The study analyzes data from 306 individuals without diabetes, focusing on insulin resistance as defined by HOMA-insulin resistance scores.
Previous studies have explored the role of gut microbiota in metabolizing nutrients in insulin resistance. This research aims to uncover the mechanisms underlying this relationship using a multi-omics approach. The study analyzes data from 306 individuals without diabetes, focusing on insulin resistance as defined by HOMA-insulin resistance scores. Until now, studies on Klotho have primarily focused on its effects on animal models such as mice, rather than directly increasing its levels in primates.
Until now, studies on Klotho have primarily focused on its effects on animal models such as mice, rather than directly increasing its levels in primates. This condition affects individuals, mostly adults over the age of 50, who physically act out their dreams during sleep, resulting in injuries to themselves or their bed partners. It's also suspected to be involved in premisses of Parkinson's disease. The study, published in the Journal of Neuroscience, presents a novel model that characterizes how REM sleep behavior disorder develops due to neurodegeneration associated with the accumulation of tau protein.
This condition affects individuals, mostly adults over the age of 50, who physically act out their dreams during sleep, resulting in injuries to themselves or their bed partners. It's also suspected to be involved in premisses of Parkinson's disease. The study, published in the Journal of Neuroscience, presents a novel model that characterizes how REM sleep behavior disorder develops due to neurodegeneration associated with the accumulation of tau protein.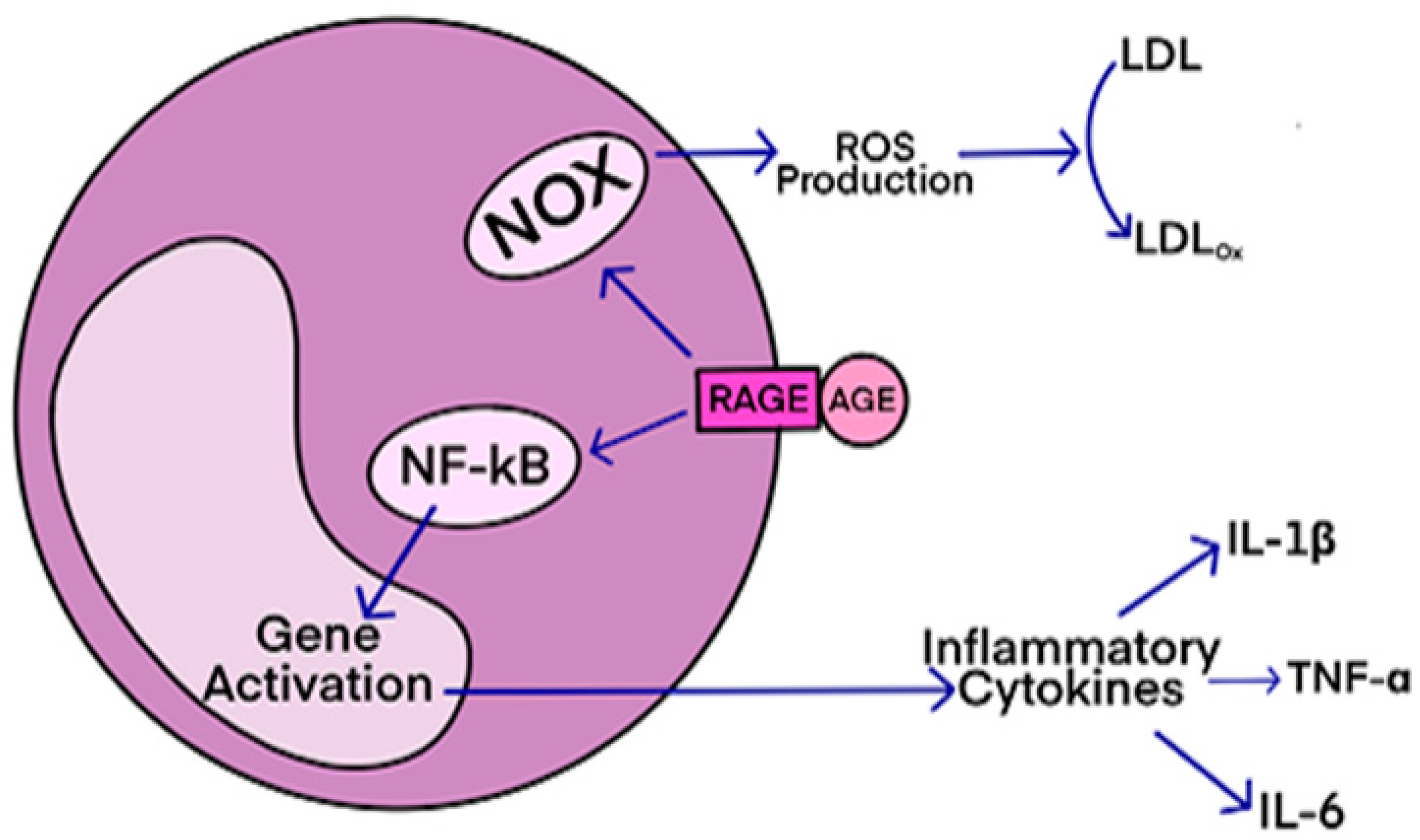 AGEs have diverse structures, but only a limited number have been characterized. Some AGEs are small molecules formed through proteolytic degradation of protein-crosslinked or protein-modified AGEs. Imbalance between the formation and destruction of AGEs, particularly under conditions of oxidative stress, results in excessive accumulation and disease progression.
AGEs have diverse structures, but only a limited number have been characterized. Some AGEs are small molecules formed through proteolytic degradation of protein-crosslinked or protein-modified AGEs. Imbalance between the formation and destruction of AGEs, particularly under conditions of oxidative stress, results in excessive accumulation and disease progression.