No one knows how to define what ALS is, its diagnosis is made by exclusion. Efforts are underway to define ALS based on molecular markers, but clearly, this would primarily be in the interest of the pharmaceutical industry.
If we do not know how to define what constitutes ALS rather than, for example, extreme cases of myasthenia gravis, it is difficult to find the cause(s).
The hypothesis of infection or poisoning is old, but apart from the case of BMAA, a neurotoxin produced by a cyanobacterium, this type of hypothesis has never been confirmed.
A peripheral hypothesis to that of infection is that of inflammation, either intraCNS or resulting from infiltration across the blood-brain barrier. There are countless studies on this subject but none are compelling.
What exactly does “inflammation” mean? Human immune systems are among the most complex systems in terms of physiology. To summarize, there are two strictly separated areas in our body: The central nervous system and the rest of the body. The CNS immune system is poorly understood. In the rest of the body, we could say that there are two kinds of subsystems, the innate nervous system which is the only one to protect us from our birth until the development of the acquired immune system. It is also the only one to protect us when the acquired immune system atrophies from the age of fifty. But these two systems interfere and the separation is only clear in medical textbooks.
Between these two systems, there is a sort of messaging system comprising many molecules with varied roles, including the famous cytokines.
One of them is called Interleukin-17A (IL-17A). A few studies have linked it to ALS since 2010, but hundreds of molecules have been associated with ALS.
 The paper we are discussing today, like most of them, does not present a major discovery, but it has some interest.
The paper we are discussing today, like most of them, does not present a major discovery, but it has some interest.
The scientists used a mouse model with a mutation in the C9orf72 ORF that impairs blood cell production. We are far from ALS, except for the C9orf72 ORF which is associated with the majority of cases of familial ALS.
They found, among other things, that this C9orf72 mutation interacts with the immune system by increasing the production of IL-17A and CD80, which is associated with autoimmune diseases such as psoriasis or multiple sclerosis.
Using an IL-17A antibody they were able to improve the condition of the mice. But usually, scientists use hyperboles very extensively. For example, one of the authors does not hesitate to say; "Our research indicates that IL-17A blockade may be quickly repurposed to treat ALS patients to slow down the progression of their disease or possibly stop ALS from ever occurring."
It's very unlikely, but who knows?
What seems surprising about this study and these claims is that the impact is not studied on the CNS immune system. The reasoning is done by analogy ("Patients with C9ORF72-related ALS similarly showed CD80 enrichment in spinal cord microglia."). Reasoning by analogy, although common in scientific publications, should be banned, you can "prove" anything this way.
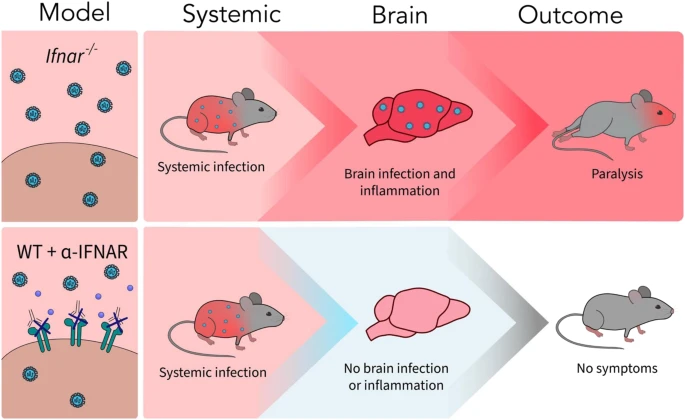 A new article aims to show that in the case of Zika viruses,
A new article aims to show that in the case of Zika viruses,  Yichang Jia, Ph.D., Co-founder of SineuGene
Yichang Jia, Ph.D., Co-founder of SineuGene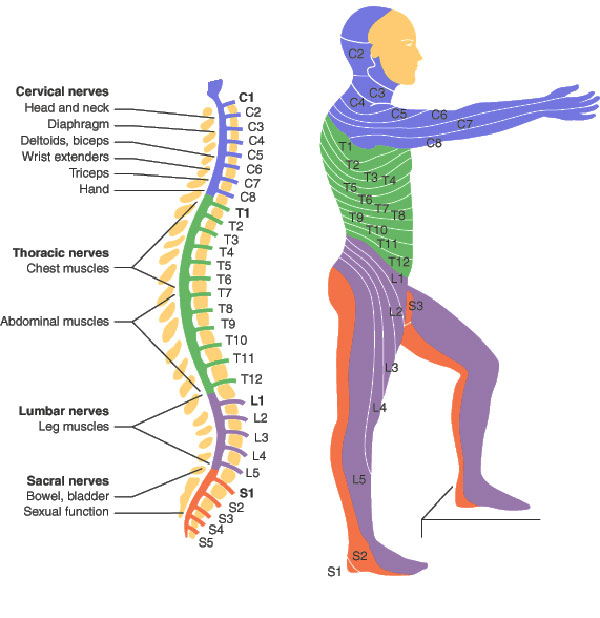 Obviously, a spinal cord injury will sever the link between the brain motor area, some upper motor neurons, and corresponding lower motor neurons and muscles. Yet it does not stop there, which is particularly interesting when we have ALS in mind. In spinal-onset ALS the disease starts in a very localized muscular for example a muscle in the thumb, and it spreads, often until respiratory muscles fail.
Obviously, a spinal cord injury will sever the link between the brain motor area, some upper motor neurons, and corresponding lower motor neurons and muscles. Yet it does not stop there, which is particularly interesting when we have ALS in mind. In spinal-onset ALS the disease starts in a very localized muscular for example a muscle in the thumb, and it spreads, often until respiratory muscles fail.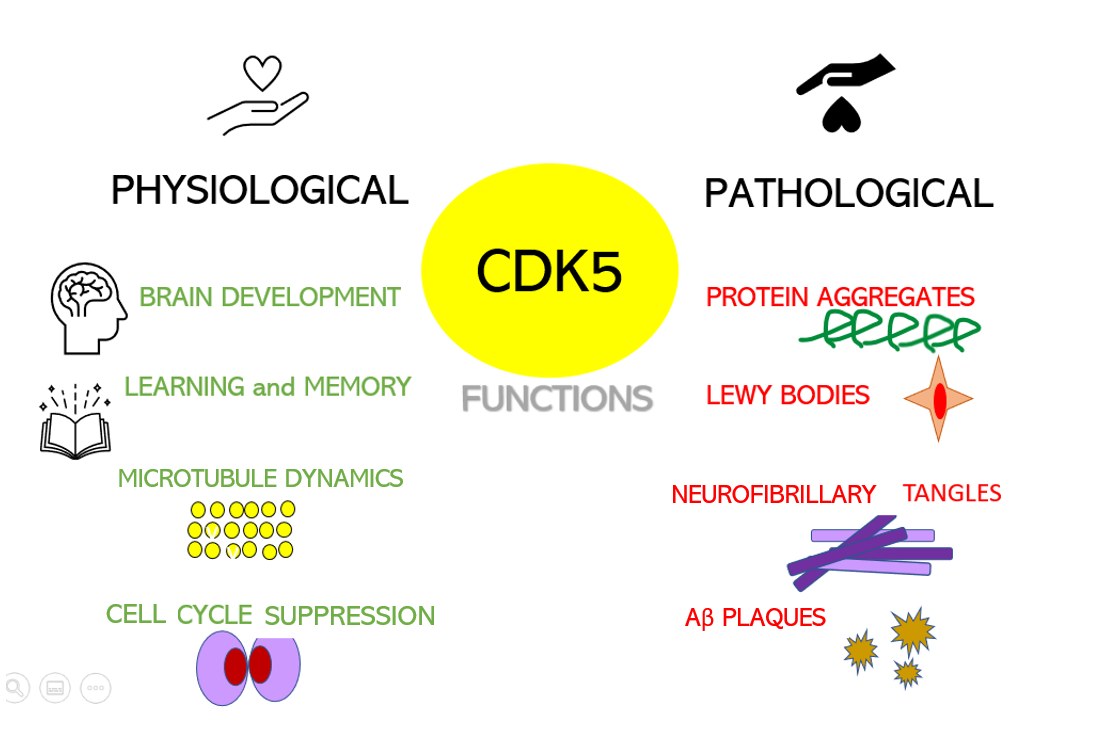 Dysregulation of CDKs, particularly cyclin-dependent kinase 5 (Cdk5), is seen in many neurological disorders, including Alzheimer's disease (AD) and Parkinson's disease (PD). Cdk5 is a unique member of the CDK family because it does not play a critical role in cell cycle progression and is not activated by a cyclin. Instead, Cdk5 is normally activated by the regulatory protein p25. Cdk5/p35/p25 activity is normally an important regulator of the proper development of the mammalian central nervous system.
Dysregulation of CDKs, particularly cyclin-dependent kinase 5 (Cdk5), is seen in many neurological disorders, including Alzheimer's disease (AD) and Parkinson's disease (PD). Cdk5 is a unique member of the CDK family because it does not play a critical role in cell cycle progression and is not activated by a cyclin. Instead, Cdk5 is normally activated by the regulatory protein p25. Cdk5/p35/p25 activity is normally an important regulator of the proper development of the mammalian central nervous system. Their results show that the model can identify, from 3 seconds of magnetoencephalographic signals, the corresponding speech segment with up to 41% accuracy, which is, however, lower than previous results. Furthermore, we know that
Their results show that the model can identify, from 3 seconds of magnetoencephalographic signals, the corresponding speech segment with up to 41% accuracy, which is, however, lower than previous results. Furthermore, we know that  If I extrapolate from Figure 6, patients with two treatments would live 13 years longer than patients without treatment. This improvement is considerable, if we ignore the statistical hallucinations of pharmaceutical companies, the current improvements simply relate to a few months of survival. We will see below that we can doubt this result.
If I extrapolate from Figure 6, patients with two treatments would live 13 years longer than patients without treatment. This improvement is considerable, if we ignore the statistical hallucinations of pharmaceutical companies, the current improvements simply relate to a few months of survival. We will see below that we can doubt this result. Any way at least if it works in human patients with FUS/C9orf72 that would mean one in five ALS patients would benefit from it. This would be much larger than Qualsody benefits.
Any way at least if it works in human patients with FUS/C9orf72 that would mean one in five ALS patients would benefit from it. This would be much larger than Qualsody benefits.