This post is about an interesting hypothesis. Hypotheses abound, yet few a convincing.
Half of patients with Alzheimer's disease, Parkinson's disease, or ALS have insulin resistance. Obesity and diabetes have been linked to neurodegenerative diseases like multiple sclerosis (MS), Alzheimer's (AD), and Parkinson's (PD). This means the cells of their body cannot let the glucose enter them. Glucose is the main energy source as it is converted into ATP. Glucose is for short-term (day) energy needs. Another source of energy is lipids (fat). Lipids are even more dense than glucose energy-wise.
The body needs an enormous amount of energy. With all the lipids in the body of a healthy person, you could charge two Tesla cars! The brain (a part of the CNS) needs 20% of all energy intake.
A new paper argues that cells shift their metabolism from glucose to lipids under stressors.
It tells that one notable distinction between glucose and lipid metabolism is in the quantity of oxygen required to generate each ATP molecule. Lipid metabolism needs two times more oxygen than glucose metabolism. The result is two times more damaging ROS (a by-product of metabolism).
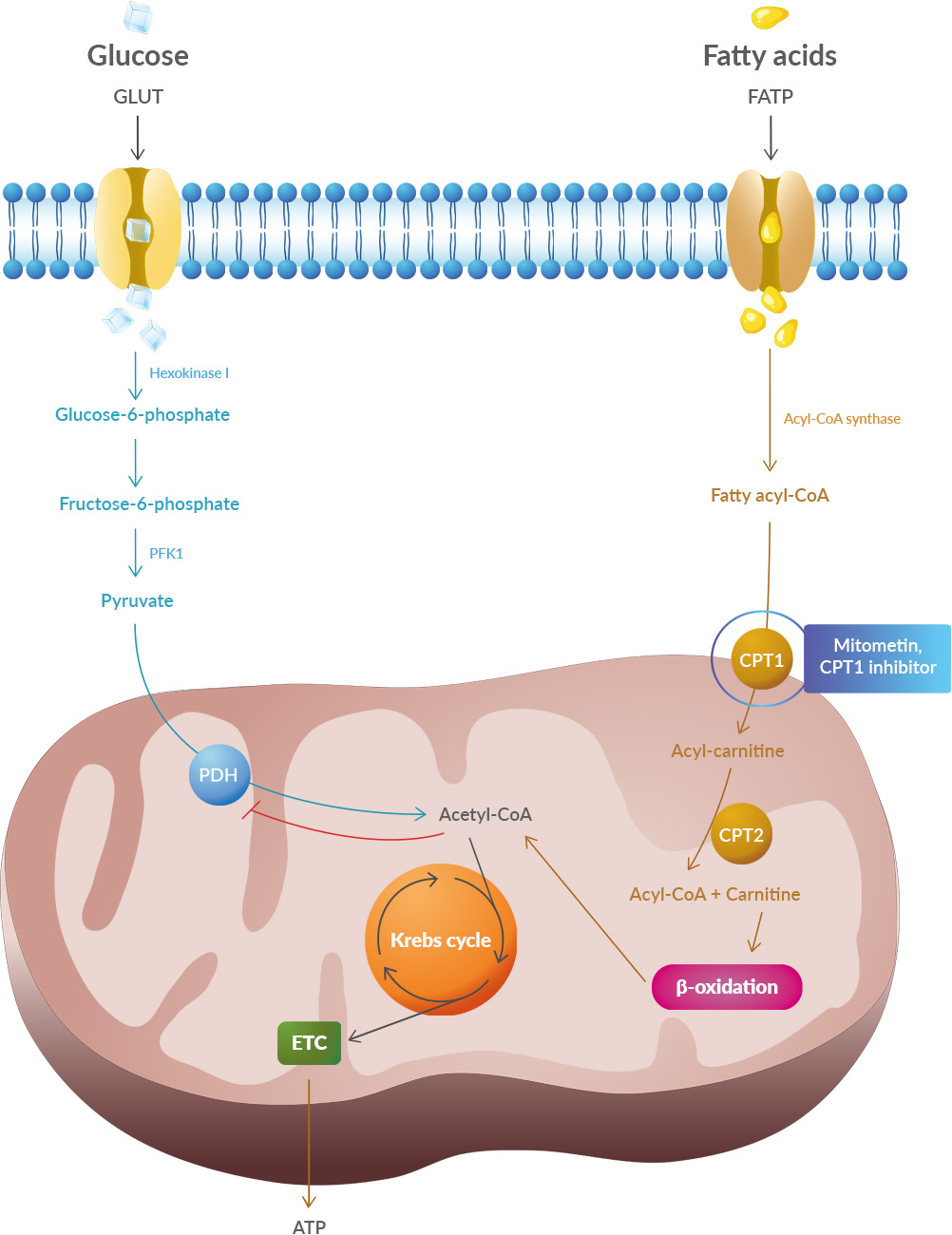 Studies have shown that oxidative stress and endoplasmic reticulum stress are correlated and can lead to protein misfolding (Abramov et al., 2020). Accumulation of misfolded proteins causes cellular damage and mitochondrial dysfunction and is associated with a range of neurodegenerative diseases, including ALS (misfolded SOD1, TDP-43, C9orf72) (McAlary et al., 2020), Parkinson's disease (misfolded α-synuclein) and Alzheimer disease (misfolded Aβ and Tau) (Abramov et al., 2020).
Studies have shown that oxidative stress and endoplasmic reticulum stress are correlated and can lead to protein misfolding (Abramov et al., 2020). Accumulation of misfolded proteins causes cellular damage and mitochondrial dysfunction and is associated with a range of neurodegenerative diseases, including ALS (misfolded SOD1, TDP-43, C9orf72) (McAlary et al., 2020), Parkinson's disease (misfolded α-synuclein) and Alzheimer disease (misfolded Aβ and Tau) (Abramov et al., 2020).
It explains also the accumulation of iron in patients' brains: To transport oxygen the blood cells need iron, and as the glucose in the blood is not absorbed in cells, it induces a change in microbiota.
It's also well known that SCFAs (including butyrate) have a positive effect on neurodegenerative diseases by their action on microbiota. SCFAs help to restore glucose as the preferred energy substrate. Authors say there are other means to restore glucose as the main source of energy.
What to think about this paper? First, some authors belong to a biotech so we can expect they want to promote their drug: Mitometin. Second, this is a review, this is not even a pre-clinical study, yet some of the authors were involved in pre-clinical studies on this topic. Other groups have written on this topic. What to make of this? Acetyl-CoA carboxylase might be of interest as they produce malonyl-CoA which inhibits the CPT1 gene that regulates lipid metabolism. B7 vitamin is known to convert acetyl-CoA to malonyl-CoA for fatty acid synthesis.

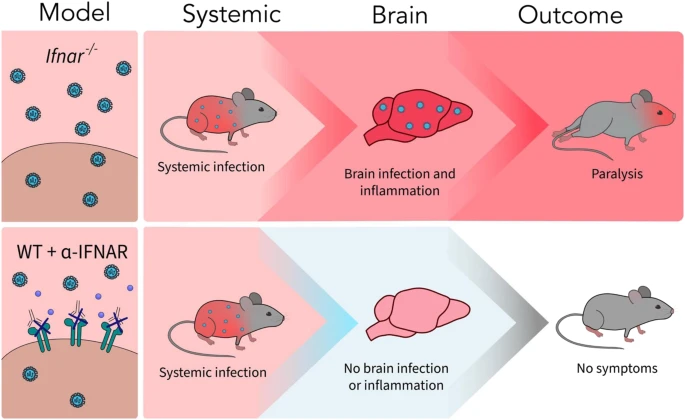 A new article aims to show that in the case of Zika viruses,
A new article aims to show that in the case of Zika viruses,  Yichang Jia, Ph.D., Co-founder of SineuGene
Yichang Jia, Ph.D., Co-founder of SineuGene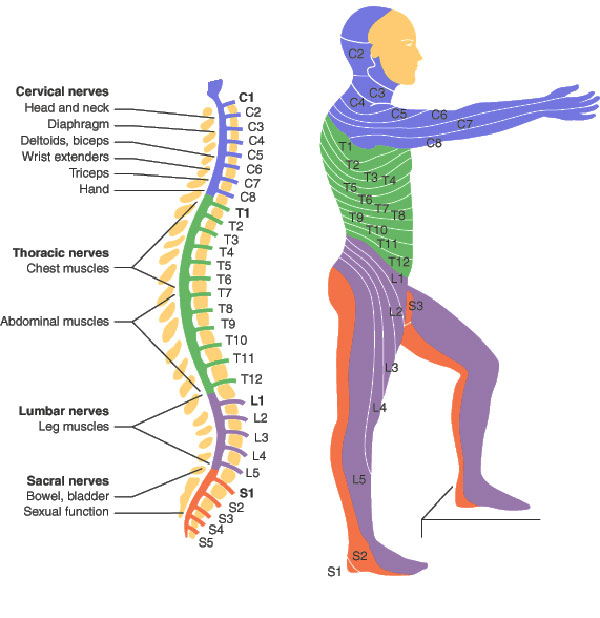 Obviously, a spinal cord injury will sever the link between the brain motor area, some upper motor neurons, and corresponding lower motor neurons and muscles. Yet it does not stop there, which is particularly interesting when we have ALS in mind. In spinal-onset ALS the disease starts in a very localized muscular for example a muscle in the thumb, and it spreads, often until respiratory muscles fail.
Obviously, a spinal cord injury will sever the link between the brain motor area, some upper motor neurons, and corresponding lower motor neurons and muscles. Yet it does not stop there, which is particularly interesting when we have ALS in mind. In spinal-onset ALS the disease starts in a very localized muscular for example a muscle in the thumb, and it spreads, often until respiratory muscles fail.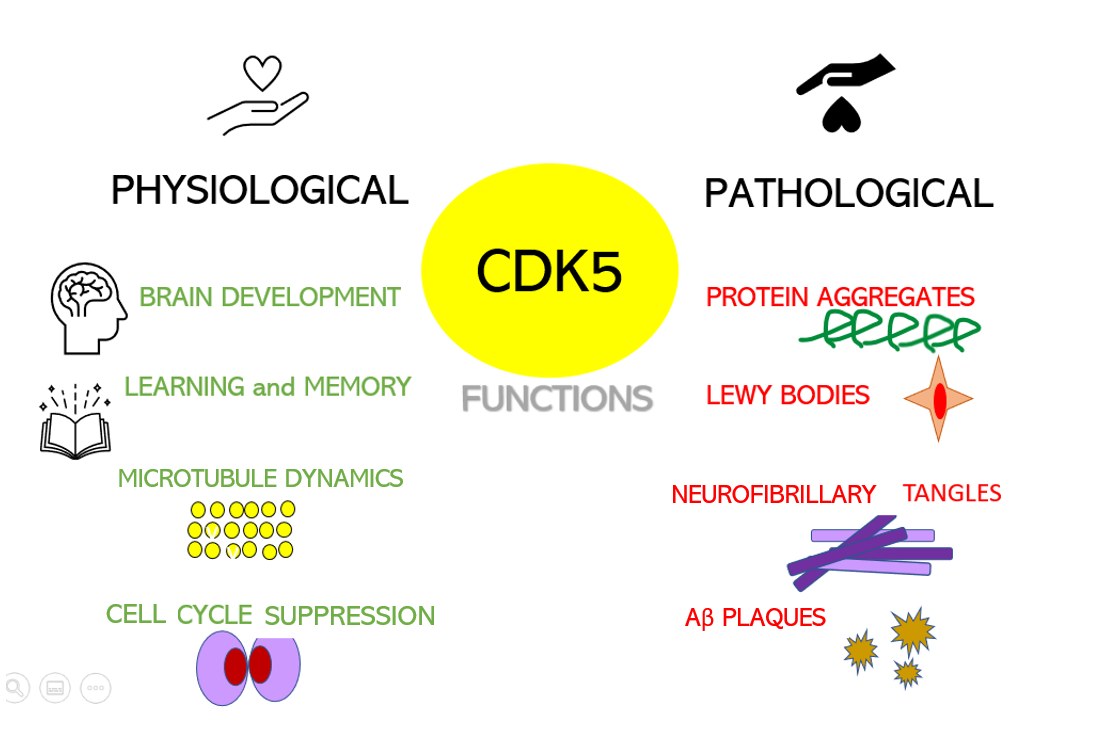 Dysregulation of CDKs, particularly cyclin-dependent kinase 5 (Cdk5), is seen in many neurological disorders, including Alzheimer's disease (AD) and Parkinson's disease (PD). Cdk5 is a unique member of the CDK family because it does not play a critical role in cell cycle progression and is not activated by a cyclin. Instead, Cdk5 is normally activated by the regulatory protein p25. Cdk5/p35/p25 activity is normally an important regulator of the proper development of the mammalian central nervous system.
Dysregulation of CDKs, particularly cyclin-dependent kinase 5 (Cdk5), is seen in many neurological disorders, including Alzheimer's disease (AD) and Parkinson's disease (PD). Cdk5 is a unique member of the CDK family because it does not play a critical role in cell cycle progression and is not activated by a cyclin. Instead, Cdk5 is normally activated by the regulatory protein p25. Cdk5/p35/p25 activity is normally an important regulator of the proper development of the mammalian central nervous system. Their results show that the model can identify, from 3 seconds of magnetoencephalographic signals, the corresponding speech segment with up to 41% accuracy, which is, however, lower than previous results. Furthermore, we know that
Their results show that the model can identify, from 3 seconds of magnetoencephalographic signals, the corresponding speech segment with up to 41% accuracy, which is, however, lower than previous results. Furthermore, we know that  If I extrapolate from Figure 6, patients with two treatments would live 13 years longer than patients without treatment. This improvement is considerable, if we ignore the statistical hallucinations of pharmaceutical companies, the current improvements simply relate to a few months of survival. We will see below that we can doubt this result.
If I extrapolate from Figure 6, patients with two treatments would live 13 years longer than patients without treatment. This improvement is considerable, if we ignore the statistical hallucinations of pharmaceutical companies, the current improvements simply relate to a few months of survival. We will see below that we can doubt this result.