Stocker une vie dans un cerveau
Comment le cerveau stocke t-il l'immense quantité d'informations auxquelles nous sommes confrontés dans un volume relativement restreint comme celui de l'hippocampe, constitue l'une des plus grandes énigmes des neurosciences. Comment le cerveau accède-t-il, sépare-t-il et organise-t-il des millions de souvenirs sans qu'ils ne se mélangent en un amas inutilisable? Et comment se fait-il que des malades d'Alzheimer oublie le nom de leur conjoint ou de leurs enfants, mais pas leur enfance?
La réponse réside dans un stockage hiérarchique et distribué par regroupement fonctionnel dynamique, où les souvenirs sont encodés non pas dans des neurones spécifiques, mais dans de vastes ensembles de neurones par le renforcement et la stabilisation dynamiques des circuits fonctionnels.
Le rôle des assemblages neuronal et du centre CA2
Le cerveau organise l'information en interconnectant des groupes fonctionnels de neurones via leurs synapses, ce que l'on appelle engrammes, qui agissent comme des « étiquettes » organisationnelles.
Ce contenu est distribué : Le contenu d’un souvenir (par exemple, les caractéristiques visuelles, la voix, les faits sémantiques) est distribué dans plusieurs aires corticales spécialisées.
L'hippocampe est une sorte de carte ou index de ces zones : L’hippocampe agit comme un index rapide et temporaire, reliant ces aires corticales distribuées pour former un souvenir cohérent.
Pour la mémoire sociale en particulier, un index crucial se situe dans une partie spécialisée de l’hippocampe appelée CA2. La région CA2 constitue le centre principal de la reconnaissance sociale dans le cerveau, encodant et maintenant la distinction entre les personnes familières et inconnues. Lorsque vous vous souvenez d’un membre de votre famille, le réseau neuronal spécifique qui représente cette personne est activé, guidé par l’index CA2.
Réseaux péri-neuronaux : Stabilisation du code social
La capacité à maintenir la netteté et la distinction de ces schémas de mémoire sociale uniques dépend fortement d’un composant structurel appelé réseau péri-neuronal.
Le réseau péri-neuronal est une structure spécialisée, dense et réticulaire, composée de la matrice extracellulaire (MEC) qui entoure les corps cellulaires et les dendrites proximales de certains neurones matures, notamment les interneurones inhibiteurs à parvalbumine (PV) à décharge rapide.
Le réseau péri-neuronal agit comme un frein moléculaire sur la plasticité synaptique, une fonction essentielle au maintien de la mémoire:
Consolidation des codes mémoriels : En se formant autour des interneurones PV dans la région CA2, les réseaux péri-neuronaux stabilisent la force synaptique des circuits qui encodent la mémoire de reconnaissance sociale. Ceci garantit la rétention à long terme de la familiarité.
Prévention des interférences : Les interneurones PV sont essentiels à l’inhibition ; ils suppriment l’activité des neurones environnants. En stabilisant ces cellules inhibitrices, les réseaux périneuronaux empêchent les interférences et maintiennent des frontières nettes et distinctes nécessaires au stockage de la mémoire.
Le lien avec la maladie d'Alzheimer
Il y a un lien connu depuis quelque temps entre les réseaux péri-neuronaux et la maladie d'Alzheimer.
L'un des symptômes les plus précoces et les plus dévastateurs de la maladie d'Alzheimer est la perte de la mémoire de reconnaissance sociale, soit la difficulté extrême à reconnaître les membres de sa famille et ses proches. La recherche a montré que ce déficit cognitif spécifique est souvent corrélé à la dégradation et à la perte des réseaux péri-neuronaux, notamment dans la région CA2 de l'hippocampe.
La théorie suggère que, lorsque les réseaux péri-neuronaux se dégradent, les interneurones PV inhibiteurs deviennent dysfonctionnels. Le « frein » moléculaire est relâché, entraînant la déstabilisation des circuits CA2. Les assemblages neuronaux distincts associés aux individus familiers ne peuvent plus être maintenus, ce qui provoque une interférence et un brouillage des codes de la mémoire sociale, rendant impossible la distinction entre les visages familiers et inconnus.
Par conséquent, le réseau péri-neuronal n'est pas un simple spectateur ; il constitue un élément clé de l'intégrité structurelle nécessaire au maintien des souvenirs spécifiques, catégorisés et vastes de notre monde social.
Réseaux péri-neuronaux ou plaques amyloïdes
De façon intéressante, certaines découvertes récentes suggèrent que la dégradation des réseaux péri-neuronaux, en particulier dans la région CA2, pourrait être un événement précoce et critique, indépendant de la formation locale de plaques amyloïdes. Ces recherches proposent que la perte de mémoire sociale dans la maladie d'Alzheimer pourrait être une conséquence directe de la dégradation des réseaux péri-neuronaux dans la région CA2, déstabilisant les circuits de la mémoire sociale, avant ou indépendamment du dépôt local et massif de plaques d'amyloïdes.
Ce point de vue complexifie la situation, suggérant que les réseaux péri-neuronaux résistent protègent les neurones qu’elles entourent des pathologies liées aux plaques, mais qu’ils finissent par être dégradées par certains processus pathologiques, ce qui entraîne une déficience cognitive importante et une progression des plaques amyloïdes.
Vers un ralentissement de la progression de la maladie Lata Chaunsali et ses collègues de Virginia School of Medicine ont utilisé le modèle murin 5XFAD de la maladie d'Alzheimer pour étudier le rôle des réseaux périneuronaux autour des neurones CA2 de l'hippocampe dans les troubles de la mémoire sociale associée à la maladie d'Alzheimer.
Les souris atteintes de la maladie d'Alzheimer présentaient une perturbation importante des réseaux péri-neuronauxs de la région CA2, associée à une altération de la mémoire sociale. L'inactivation génétique ou enzymatique des réseaux péri-neuronaux de la région CA2 chez les souris saines reproduisait ces altérations. L'analyse transcriptomique révèle une surexpression des métalloprotéinases matricielles (MMP) clivant les réseaux péri-neuronaux chez les souris atteintes de la maladie d'Alzheimer, induisant un déséquilibre entre la synthèse et le remodelage des réseaux péri-neuronaux.
Les auteurs ont alors testé si les inhibiteurs de MMP — une classe de médicaments déjà étudiée pour leur potentiel dans le traitement du cancer et de l'arthrite — pouvaient prévenir la perte des réseaux périneuronaux. L'ilomastat (code de développement GM6001 et nom commercial Galardin) est un inhibiteur de métalloprotéinases matricielles à large spectre.
Le traitement s'est avéré plutôt efficace, empêchant d'autres lésions et aidant les souris à conserver leurs souvenirs les unes des autres. L'inhibition de l'activité des métalloprotéinases matricielles (MMP) par le GM6001 a prévenu la perturbation des réseaux péri-neuronaux et protègé contre les déficits de mémoire sociale dans ce modèle murin de maladie d'Alzheimer. Évidemment il s'agit là d'une avancée vers un médicament qui ralentirait la progression de la maladie, mais les dégâts étant déjà présents, il est impossible de restaurer les souvenirs disparus.
Une interrogation demeure sur la cause de la destruction des réseaux péri-neuronaux par les métalloprotéinases. En général cela arrive à la suite d’évènements stressants imposant un remodelage des tissus. La recherche a donc des pistes intéressantes à creuser.
Globalement, cette étude fournit également des éléments convaincants que l'inhibition pharmacologique de la protéolyse du réseau péri-neuronal peut prévenir la perte de mémoire sociale, suggérant que les réseaux péri-neuronaux constituent une cible thérapeutique potentielle.

 By Manu5 - http://www.scientificanimations.com/wiki-images/
By Manu5 - http://www.scientificanimations.com/wiki-images/ Ils ont confirmé qu'une exposition prolongée aux NRTI était associée à un risque moindre de développer la maladie d’Alzheimer.
- D’après les données du VA, chaque année supplémentaire de traitement par NRTI était associée à une réduction de 4 à 6 % du risque de maladie d’Alzheimer.
- D’après les données de MarketScan, la réduction était encore plus marquée : de 10 à 13 % par année d’utilisation.
Ils ont confirmé qu'une exposition prolongée aux NRTI était associée à un risque moindre de développer la maladie d’Alzheimer.
- D’après les données du VA, chaque année supplémentaire de traitement par NRTI était associée à une réduction de 4 à 6 % du risque de maladie d’Alzheimer.
- D’après les données de MarketScan, la réduction était encore plus marquée : de 10 à 13 % par année d’utilisation.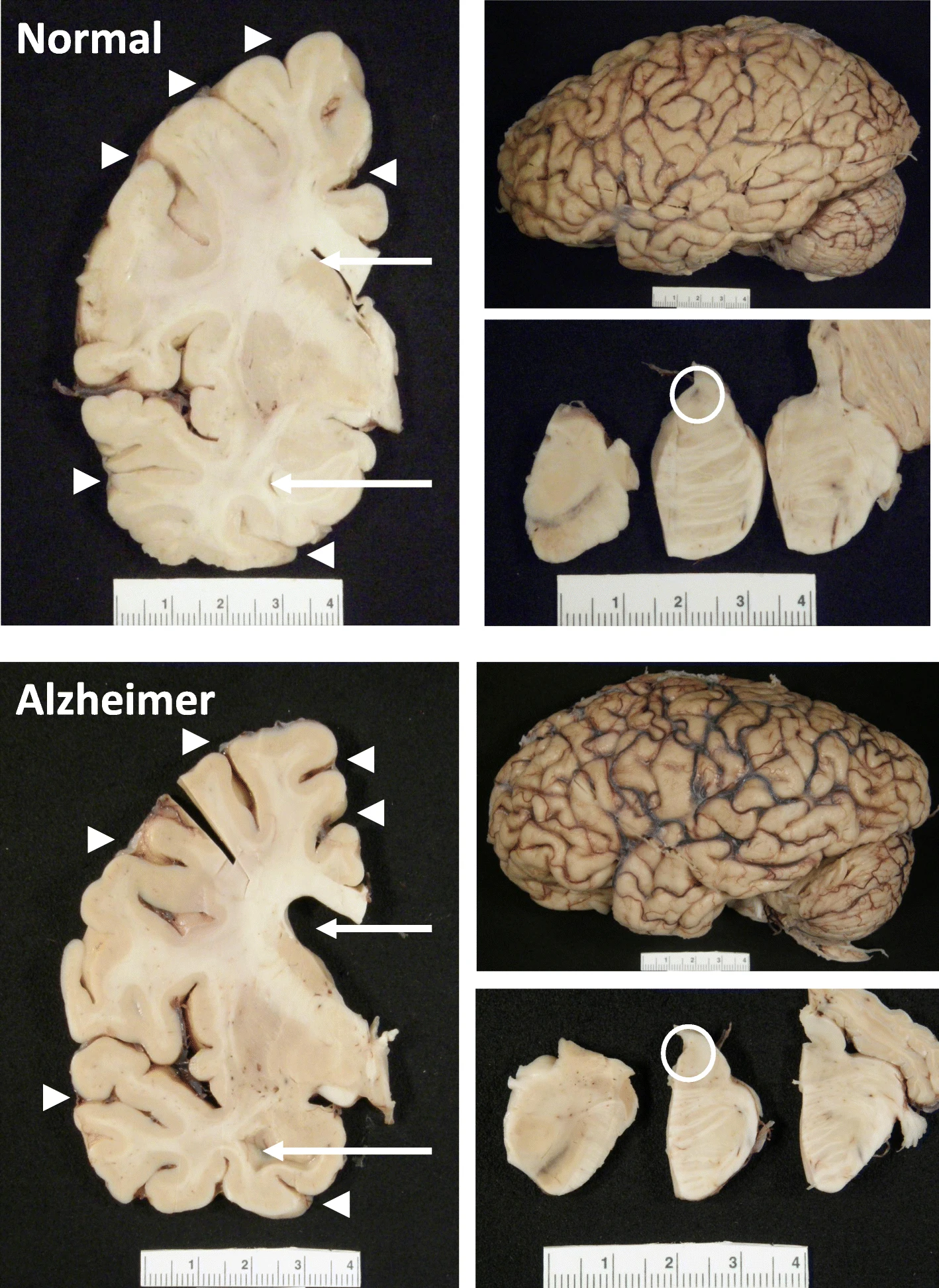 The hippocampus, a small, seahorse-shaped structure buried deep within the brain, is best known for its role in memory formation and learning. It is an exceptionally vulnerable structure, with perfusion deficits often observed in diseases related to learning and memory. However, a brain affected by Alzheimer's disease tends to exhibit at least moderate cortical atrophy, including in the precuneus and posterior cingulate gyrus. It should be noted that the posterior cingulate gyrus is adjacent to the hippocampus.
The hippocampus, a small, seahorse-shaped structure buried deep within the brain, is best known for its role in memory formation and learning. It is an exceptionally vulnerable structure, with perfusion deficits often observed in diseases related to learning and memory. However, a brain affected by Alzheimer's disease tends to exhibit at least moderate cortical atrophy, including in the precuneus and posterior cingulate gyrus. It should be noted that the posterior cingulate gyrus is adjacent to the hippocampus.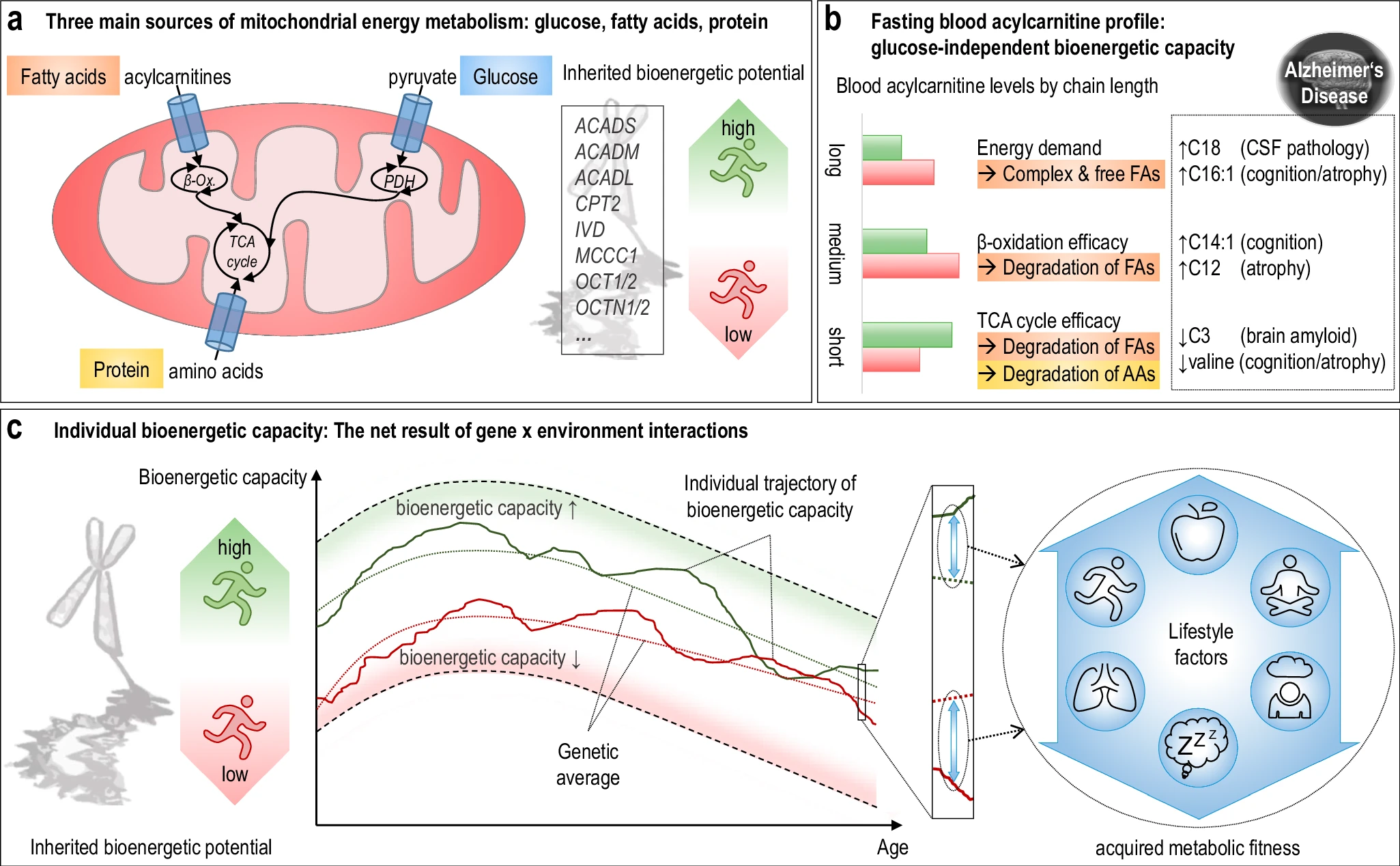 The researchers used acylcarnitine profiles from blood samples to identify distinct bioenergetic subgroups in Alzheimer's Disease (AD) patients and evaluate how bioenergetic capacity relates to disease progression.
They used data from 1,531 participants in the Alzheimer's Disease Neuroimaging Initiative (ADNI), and identified several bioenergetic subgroups with significant differences in AD biomarkers, cognitive function, and brain glucose metabolism.
These subgroups were primarily determined by modifiable factors (40-60%) related to beta-oxidation function, rather than genetic factors, suggesting potential for intervention.
The researchers used acylcarnitine profiles from blood samples to identify distinct bioenergetic subgroups in Alzheimer's Disease (AD) patients and evaluate how bioenergetic capacity relates to disease progression.
They used data from 1,531 participants in the Alzheimer's Disease Neuroimaging Initiative (ADNI), and identified several bioenergetic subgroups with significant differences in AD biomarkers, cognitive function, and brain glucose metabolism.
These subgroups were primarily determined by modifiable factors (40-60%) related to beta-oxidation function, rather than genetic factors, suggesting potential for intervention. Alzheimer's disease research has produced many hypotheses over the years, including cholinergic, inflammatory, viral, mitochondrial, tau, and amyloid. However, none of these hypotheses have led to treatments that can stop or reverse the disease. This leads to a search for new theories to explain these failures. But this may be because interventions occur too late in the disease progression, with brain damage irreparable and compensatory mechanisms saturated.
Alzheimer's disease research has produced many hypotheses over the years, including cholinergic, inflammatory, viral, mitochondrial, tau, and amyloid. However, none of these hypotheses have led to treatments that can stop or reverse the disease. This leads to a search for new theories to explain these failures. But this may be because interventions occur too late in the disease progression, with brain damage irreparable and compensatory mechanisms saturated.