Mounir El Maï, Marta Marzullo, Inês Pimenta de Castro, Miguel Godinho Ferreira
Here is an interesting article from the point of view of neurodegenerative diseases. There are terms that resonate with those we are familiar with: ROS, SOD, insulin resistance. Although this article does not apply to neurons (as they do not divide) it could provide a key to ALS and other neurodegenerative diseases.
 * Source: https://elifesciences.org/articles/54935/figures*
* Source: https://elifesciences.org/articles/54935/figures*
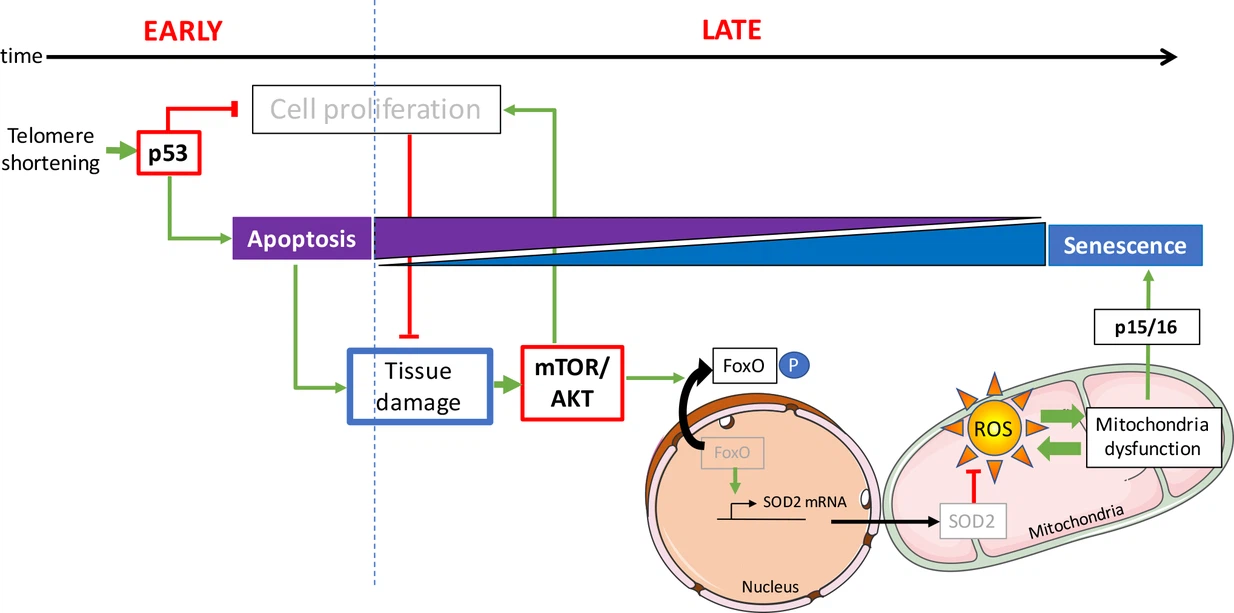
Indeed it presents the idea that in a young organism, a defective cell commits suicide and is easily replaced, while in an aged organism a defective cell seeks to subsist despite everything, and that damages the tissue where it is inserted. The determining factor for this change in behavior is the quality control of DNA.
Whenever a chromosome in a eukaryotic cell is replicated, DNA polymerase is unable to copy the last nucleotides. To overcome this problem, the DNA of a chromosome is equipped with a telomer at each end. It is a DNA region at the end of a chromosome, which is without genetic information. During each cell division, the telomeres erode until reaching a critical size.
Stopping the cell cycle allows the cell to stimulate its repair mechanisms, some of which are directly activated by the p53 pathway. When the repair is done, the p53 level returns to normal and the cell cycle resumes. If the damage to the cell cannot be repaired, the cell goes into apoptosis, which leads to its elimination.
Cell proliferation is however possible by inactivation of the p53 pathway. Cells entering proliferation after inactivation of the p53 pathway undergo gross chromosomal rearrangements and genome instability.
In this article, the authors study the effect of reducing telomeres in a model animal (zebrafish) where this reduction is accelerated.
In most eukaryotes, shortening of telomeres is counteracted by telomerase, although its expression is limited in most human somatic cells. As a result, telomeres shorten considerably during human aging. But, and this is not explained in the article, this is only true for cells that are subject to frequent replacement.
Curiously, while neurodegenerative diseases are linked to age, neurons are not subject to the Hayflick limit, even if there is one for glial cells. Perhaps the mechanism described in this article induces neurodegenerative diseases by its action on glial cells ?.
One may wonder to what extent the senescence of neurons and other glial cells leads to proteopathies. For example, if there is not enough energy, the endoplasmic reticulum will not be able to fulfill its role of protein conformation, and similarly the Golgi apparatus will not be able to deliver these proteins to their normal destination. This could lead to proteopathy.
 * Source: Magnus Manske via Wikipedia *
* Source: Magnus Manske via Wikipedia *
But what motivates this article is that the molecular mechanism determining cell fate (apoptosis or senescence) remains uncertain.
In the present study, the authors describe young zebrafish (3 months old), which are deficient in telomerase already show DNA damage and activation of p53. At this stage, apoptosis is the predominant cellular fate. Even though DNA damage is present in proliferative tissues, such as the intestine and testes, no signs of cellular senescence could be detected. In this study, early activation of p53 in the zebrafish does not affect the function of the mitochondria.
However, there is an obvious change between apoptosis and senescence in older fish. In these animals, senescence becomes the most widespread cellular response. This observation highlights the fact that the same types of cells can undergo different cellular destinies in vivo depending on the age of the animal.
Unlike young fish, the gut and testes of older zebrafish exhibit mitochondrial dysfunction accompanied by a significant reduction in ATP levels (ATP provides the energy necessary for the chemical reactions of cellular metabolism) and an accumulation of ROS (while they are byproducts of normal oxygen metabolism, their concentration can increase significantly in times of stress and damage cell structures).
The study reveals that mitochondrial defects are associated with a reduction in mitochondrial defenses. The authors observe that with age, the expression of SOD2 is reduced in response to activation of AKT pathway.
In humans, there are 3 genes from the Akt family: Akt1, Akt2, and Akt3. These enzymes belong to the family of protein kinases. Protein kinase B (PKB), also known as Akt, is a serine/threonine-specific protein kinase that plays a key role in several cellular processes such as glucose metabolism, apoptosis, cell proliferation, transcription and cell migration. AKT is activated on pro-proliferative extracellular signals. The mTOR / AKT pathway is triggered by growth factor receptors, including the insulin growth factor receptor (IGFR).
The anti-proliferative p53 and pro-survival mTOR / AKT pathways interact in complex ways. Depending on the context, the interaction of these pathways modulates cell fate by stopping the cell cycle, apoptosis or senescence. Consistent with these in vitro data, scientists show that activation of AKT in aged mutant zebrafish is concomitant with suppression of apoptosis and activation of mechanisms leading to senescence.
What is the mechanism that switches the cell fate from apoptosis to senescence? A high rate of apoptosis among tissue cells increases the demand for cell proliferation of surrounding cells in a process called compensatory proliferation induced by apoptosis.
On the contrary, the restrictions on cell proliferation cause tissue degeneration which becomes apparent in aging zebrafish.
In tissues where stem cells are not readily available or where tissue-intrinsic genetic programs limit cell division, cell hypertrophy is an alternative strategy to tissue homeostasis.
