Another interesting article was published by Alzforum.
Alzforum is a quality news website dedicated to Alzheimer’s disease and other neurodegenerative disorders. It is a subsidiary Fidelity Management & Research.
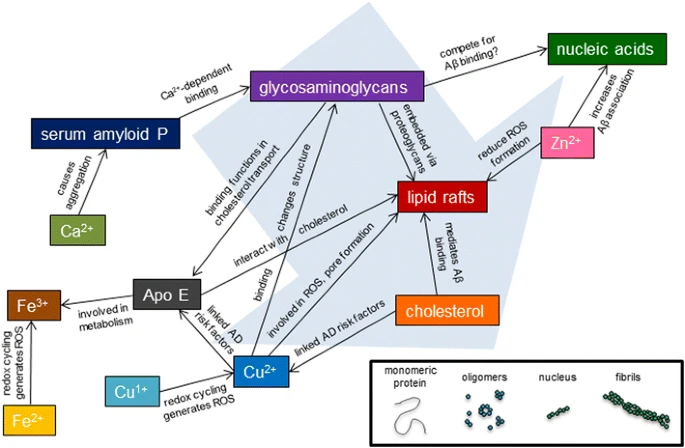 In Alzheimer disease, aggregation of Aβ42 peptide into amyloids is conceived as the pathogenic trigger of a cascade leading to tau accumulation into neurofibrillary tangles, neuronal loss, and clinical dementia. However, while most of the 40 anti-amyloid clinical trials over the past two decades have successfully reduced the burden of brain amyloid, corresponding benefits for the patients have never materialized.
In Alzheimer disease, aggregation of Aβ42 peptide into amyloids is conceived as the pathogenic trigger of a cascade leading to tau accumulation into neurofibrillary tangles, neuronal loss, and clinical dementia. However, while most of the 40 anti-amyloid clinical trials over the past two decades have successfully reduced the burden of brain amyloid, corresponding benefits for the patients have never materialized.
Moreover, brain amyloidosis does not invariably predict dementia: by the age of 85, the prevalence of brain amyloidosis is approximately 60% whereas that of dementia is only of 10%.
This new study makes the revolutionary hypothesis that high levels of natively-folded, soluble Aβ42 are associated with normal cognition in the setting of brain amyloidosis.
In a cross-sectional analysis of 598 brain amyloid-positive individuals participating in the Alzheimer's Disease Neuroimaging Initiative, higher levels of soluble Aβ42 were associated with normal cognition.
Higher soluble Aβ42 levels were also associated with better neuropsychological performance and larger hippocampal volume, with a larger effect size yielded by changes in soluble Aβ42 than in insoluble (brain amyloid) Aβ42.
“The main premise on which Alzheimer’s and all neurodegenerative diseases are conceived, is essentially the idea that proteins are toxic. It should end,” Alberto Espay, University of Cincinnati, told Alzforum.
Espay and Ezzat want their findings to inspire a paradigm shift on how we view neurodegenerative disease. “Our key message is that neurodegenerative diseases, in general, are associated with loss of protein,” said Espay. He contends that yes, aggregates accumulate, but total soluble protein goes down and that is what leads to disease. Tau protein levels falls in tauopathies, as synuclein in falls in Parkinson’s, Aβ in Alzheimer's disease, and progranulin in FDD/ALS.
The situation in Parkinson's disease mirrors what the scientists found in Alzheimer's disease. Most cases of Parkinson's disease have no specific known cause. A small proportion of cases, however, can be attributed to known genetic factors. Environmental toxins, herbicides, pesticides, and fungicides, as well as some medical and recreational drugs have been associated with the risk of developing PD. Vascular events such as stroke can cause Parkinson's disease. As for ALS, there are many conditions that look similar to Parkinson's disease. The motor symptoms of the disease result from the death of cells in the substantia nigra, a region of the midbrain.
For several generations of neurologists, the alpha-synuclein protein has been at the center of the Parkinson's disease universe. Alpha-synuclein is exists in the same form since prehistoric genomes. While the function of a protein molecule generally depends on its correct shape, wouldn't adopting an “incorrectly shaped” beta sheet aggregate make it impossible for it to function?
The central event was the discovery in 1997 that autosomal dominant Parkinson's disease was caused by a point mutation in the SNCA gene. Alpha-synuclein aggregates to form insoluble fibrils in pathological conditions characterized by Lewy bodies, such as Parkinson's disease, dementia with Lewy bodies and multiple system atrophy.
The elegant work of Braak and colleagues on the brains of patients under 50 with Parkinson's disease has shown that alpha-synuclein aggregates in a stereotypical pattern, conspicuously first appearing in the peripheral nervous system, then into the central nervous system.
As with the beta-amyloid protein in Alzheimer's disease, the elimination of alpha-synuclein in young mice makes no difference and actually protects them from the effects of MPTP, a mitochondrial toxin. Surprisingly, knockout mice, where the SNCA gene has been turned off, develop deficits when they get old!
One of the curious things about Lewy bodies is that the proportion of substantia nigra neurons containing Lewy pathology remains relatively constant regardless of how many neurons are already lost, which invalidates the classic belief that it is Lewy bodies that cause cell death in the substancia nigra.
Is it really the higher level of proteins, normal or mutated, that ultimately leads to neurodegenerative diseases?
Advertisement

This book retraces the main achievements of ALS research over the last 30 years, presents the drugs under clinical trial, as well as ongoing research on future treatments likely to be able stop the disease in a few years and to provide a complete cure in a decade or two.
 Source: KieranMaher at English Wikibooks
Source: KieranMaher at English Wikibooks
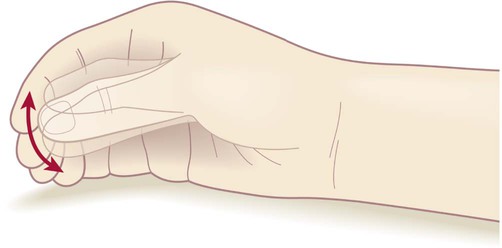 ‘Pill-rolling’ rest tremor as found in Parkinson’s disease.
‘Pill-rolling’ rest tremor as found in Parkinson’s disease.
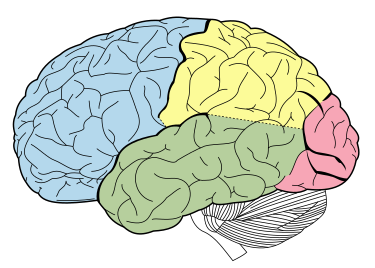 Source: Wikipedia. The temporal lobe is shown in green, while the motor area is in the frontal lobe in blue
Source: Wikipedia. The temporal lobe is shown in green, while the motor area is in the frontal lobe in blue Hagmann P, Cammoun L, Gigandet X, Meuli R, Honey CJ, et al.
Hagmann P, Cammoun L, Gigandet X, Meuli R, Honey CJ, et al.  In silico computer models for DBS allow to pre-select a set of potentially optimal stimulation parameters. If efficacious, they could further carry insight into the mechanism of action of DBS and foster the development of more efficient stimulation approaches. In recent years, the focus has shifted towards DBS-induced firing in myelinated axons, deemed particularly relevant for the external modulation of neural activity.
In silico computer models for DBS allow to pre-select a set of potentially optimal stimulation parameters. If efficacious, they could further carry insight into the mechanism of action of DBS and foster the development of more efficient stimulation approaches. In recent years, the focus has shifted towards DBS-induced firing in myelinated axons, deemed particularly relevant for the external modulation of neural activity.