Here is a summary of an educational article that will be of particular interest to people affected by ALS (Lou Gehrig in the USA/Charcot's disease in France).
Essentially the article explains that ALS is a disease specific to certain nerve formations that are absent in most mammals, except certain higher primates (and perhaps other mammals like the Degu). These formations being absent in rats and mice, we wonder what the purpose of preclinical trials on these rodents is. I think this is the main contribution of the article.
There are indeed key differences in the organization and function of the corticospinal system in primates compared to non-primates, such as rodents.
 Cortico-motoneuronal projection is a late evolutionary development that is present only in dexterous primates, such as capuchins, macaques, great apes, and humans, but absent in adult rodents, carnivores, and many others. primates, such as marmosets.
Cortico-motoneuronal projection is a late evolutionary development that is present only in dexterous primates, such as capuchins, macaques, great apes, and humans, but absent in adult rodents, carnivores, and many others. primates, such as marmosets.
Although tool use is not exclusive to advanced primates and humans with demonstrable cortico-motoneuronal connections, primates exhibit a wide range of tool-making and use behaviors. Why did the direct projection from the cortex to the α motor neuron appear so late in evolution? One possibility is that the cortico-motoneuronal system acts selectively on the motor apparatus of the upper limb to allow relatively independent finger movements. These movements are essential for performing all skilled manual tasks, including key human motor characteristics, such as gestures and tool use.
Corticospinal neurons located in the motor areas of the cerebral neocortex project axons onto the spinal network. The corticospinal system of primates has a wider cortical origin than in other animals and a wide range of fiber diameters, including thick, rapidly conducting axons. Direct cortico-motoneuronal projections from the motor cortex to motor neurons of the arm and hand are a recent evolutionary feature, well developed in dexterous primates and particularly in humans. This system is involved in the control of skilled movements, performed with the splitting of the distal extremities and at low levels of force. During movement, corticospinal neurons are activated in a very different way from “lower” motor neurons, and there is no simple or fixed functional relationship between a so-called “upper” motor neuron and its target lower motor neuron, whereas in other mammals there is an interneuron which makes the junction between an upper motor neuron and a lower motor neuron.
During the development of ALS, there is a selective loss of rapidly conducting corticospinal axons and their synaptic connections, which is reflected in responses to noninvasive cortical stimuli and measures of corticomuscular coherence.
A given muscle can be used in different ways, and one of the key features of the cortico-motoneuronal system is the recruitment of particular, task-specific muscle groups. There is task-specific flexibility between the activity of a cortico-motoneuron and its target motoneurons. This is lost when the cortico-motoneuronal projection is dysfunctional and, as would be expected, there will then be deficits in skill as well as muscle strength.
The well-known weakness and loss of motor units in the hand muscles of ALS patients is more pronounced in the thenar (thumb) muscles than in the hypothenar (little finger) muscles. Although several different factors are known to contribute to lower motor neuron (spinal) dysfunction, these particular changes are due to the greater loss of the cortico-motoneuronal system on the thumb muscles compared to the little finger muscles.
This particular vulnerability of muscle groups heavily used during ALS is not limited to the hand. This includes studies on “split elbow,” “split foot,” and “split ankle.” In all three syndromes, the most profound weakness was seen in muscle groups that, in healthy controls, are known to receive relatively strong cortico-motoneuronal projections.
Along with the corticospinal projection from the cortex to the spinal cord, there is a significant corticobulbar projection to the motor centers of the brainstem, which is essential for actions such as speaking, chewing, and swallowing.
It is important to clarify that the corticospinal system is multifunctional and concerns not only movement but it also has somatosensory, autonomic, and trophic functions. When preparing for and executing the movement, it does not work in isolation, but in concert with other motor systems in the brainstem and spine.
In conclusion, because in rodents, corticospinal projections from the sensorimotor cortex primarily avoid the ventral horn and have limited direct effects on motor control, the pyramidal neurons giving rise to these projections are not considered similar "higher motor neurons." to those of primates, it is therefore not a good model for studying the effects of potential drugs on ALS. A small number of carefully designed studies in higher primates remain needed to advance the understanding and treatment of ALS.
 What is interesting is that 14-3-3θ seems to interact preferentially with pathogenic TDP-43 versions but not with the usual version of TDP-43. This suggests that reducing the production (or increasing the degradation) of 14-3-3θ would reduce the production of pathogenic TDP-43. Scientists have therefore sought to reduce the amount of this 14-3-3θ protein in cells through genetic therapy.
What is interesting is that 14-3-3θ seems to interact preferentially with pathogenic TDP-43 versions but not with the usual version of TDP-43. This suggests that reducing the production (or increasing the degradation) of 14-3-3θ would reduce the production of pathogenic TDP-43. Scientists have therefore sought to reduce the amount of this 14-3-3θ protein in cells through genetic therapy.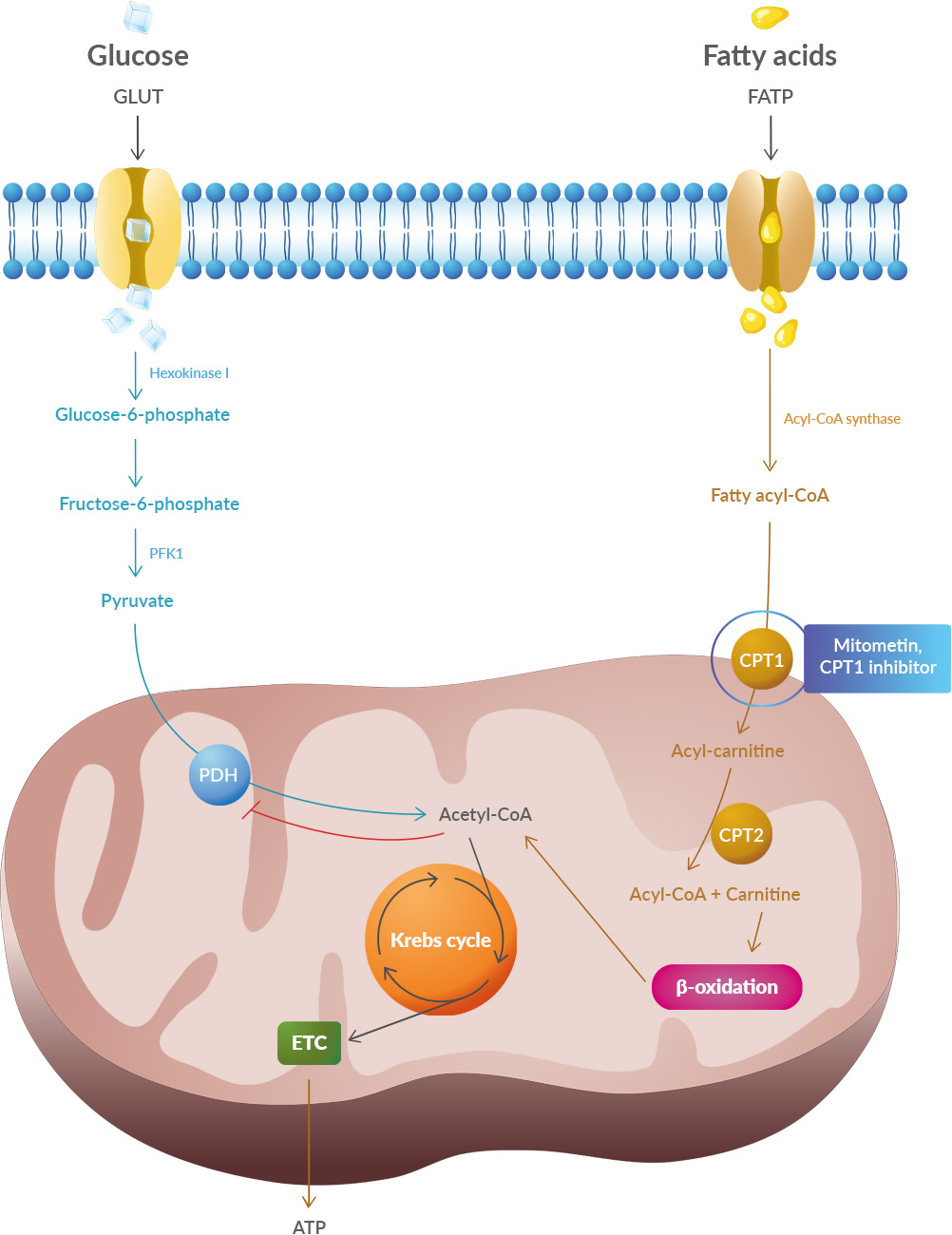 Studies have shown that oxidative stress and endoplasmic reticulum stress are correlated and can lead to protein misfolding (Abramov et al., 2020). Accumulation of misfolded proteins causes cellular damage and mitochondrial dysfunction and is associated with a range of neurodegenerative diseases, including ALS (misfolded SOD1, TDP-43, C9orf72) (McAlary et al., 2020), Parkinson's disease (misfolded α-synuclein) and Alzheimer disease (misfolded Aβ and Tau) (Abramov et al., 2020).
Studies have shown that oxidative stress and endoplasmic reticulum stress are correlated and can lead to protein misfolding (Abramov et al., 2020). Accumulation of misfolded proteins causes cellular damage and mitochondrial dysfunction and is associated with a range of neurodegenerative diseases, including ALS (misfolded SOD1, TDP-43, C9orf72) (McAlary et al., 2020), Parkinson's disease (misfolded α-synuclein) and Alzheimer disease (misfolded Aβ and Tau) (Abramov et al., 2020).
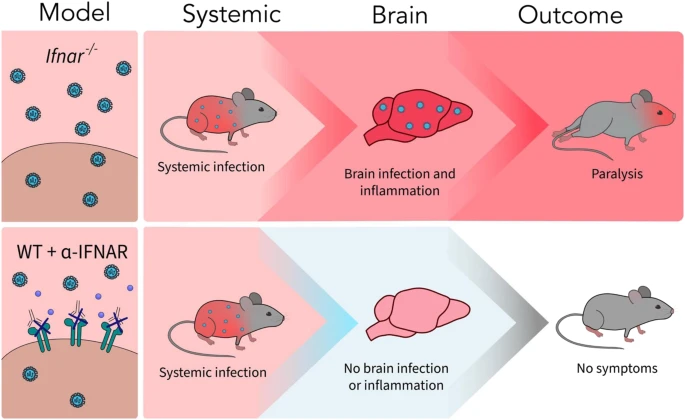 A new article aims to show that in the case of Zika viruses,
A new article aims to show that in the case of Zika viruses,  Yichang Jia, Ph.D., Co-founder of SineuGene
Yichang Jia, Ph.D., Co-founder of SineuGene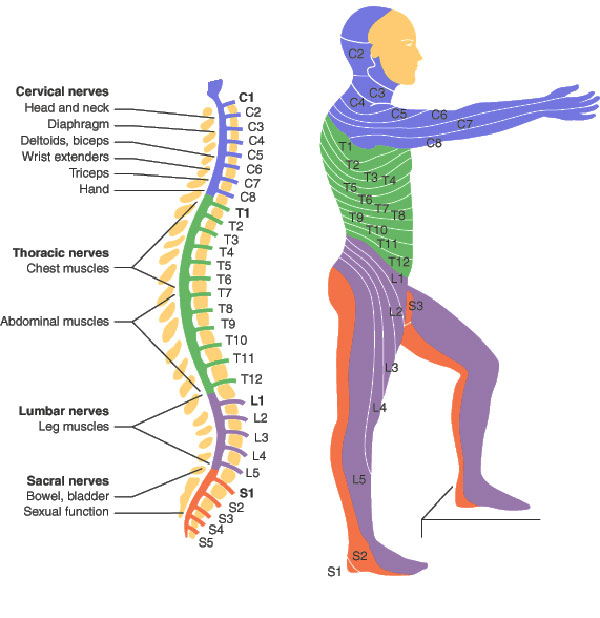 Obviously, a spinal cord injury will sever the link between the brain motor area, some upper motor neurons, and corresponding lower motor neurons and muscles. Yet it does not stop there, which is particularly interesting when we have ALS in mind. In spinal-onset ALS the disease starts in a very localized muscular for example a muscle in the thumb, and it spreads, often until respiratory muscles fail.
Obviously, a spinal cord injury will sever the link between the brain motor area, some upper motor neurons, and corresponding lower motor neurons and muscles. Yet it does not stop there, which is particularly interesting when we have ALS in mind. In spinal-onset ALS the disease starts in a very localized muscular for example a muscle in the thumb, and it spreads, often until respiratory muscles fail.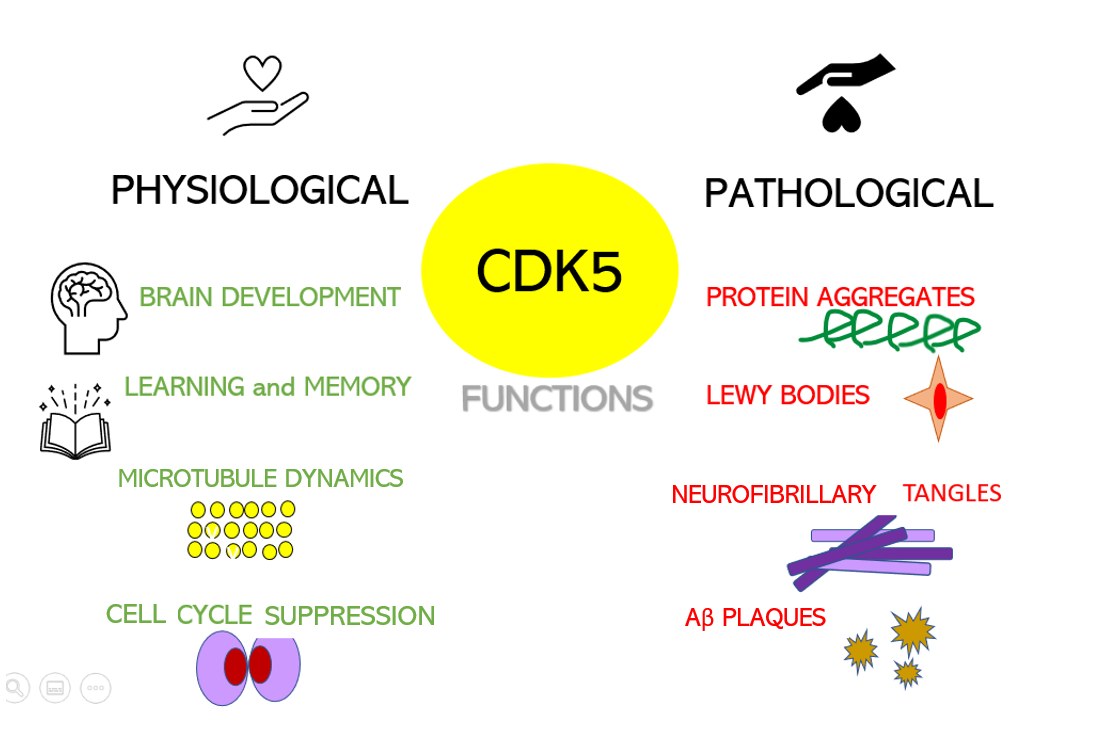 Dysregulation of CDKs, particularly cyclin-dependent kinase 5 (Cdk5), is seen in many neurological disorders, including Alzheimer's disease (AD) and Parkinson's disease (PD). Cdk5 is a unique member of the CDK family because it does not play a critical role in cell cycle progression and is not activated by a cyclin. Instead, Cdk5 is normally activated by the regulatory protein p25. Cdk5/p35/p25 activity is normally an important regulator of the proper development of the mammalian central nervous system.
Dysregulation of CDKs, particularly cyclin-dependent kinase 5 (Cdk5), is seen in many neurological disorders, including Alzheimer's disease (AD) and Parkinson's disease (PD). Cdk5 is a unique member of the CDK family because it does not play a critical role in cell cycle progression and is not activated by a cyclin. Instead, Cdk5 is normally activated by the regulatory protein p25. Cdk5/p35/p25 activity is normally an important regulator of the proper development of the mammalian central nervous system.